Medizinische/ Biologische Chemie
Untersuchungen von Inhibitionsmechanismen kovalenter Inhibitoren
Aufgrund von vielfältigen Vorteilen erfahren kovalente Liganden zur Zeit eine Renaissance nicht nur in der Wissenschaft, sondern auch im industriellen Kontext.1 Bezüglich ihrer Verwendung als Arzneistoffe sind sie in vivo effizienter und können in geringen Dosen und mit reduzierter Häufigkeit aufgrund verlängerter residence time am Target verabreicht werden.
Zusätzlich bietet das Konzept der kovalenten Modifikation von Proteinen weitere Möglichkeiten Targets zu adressieren, welche durch niedermolekulare Wirkstoffe als nicht adressierbar angenommen wurden, da spezifische Bindungstaschen nicht vorhanden waren („drug the undruggable“) 2. Bisher jedoch wurden die meisten kovalenten Liganden zufällig gefunden, nicht nur weil Werkzeuge für die rationale Optimierung oder de novo Design nicht ausreichend etabliert und ausgearbietet wurden, in Relation zu den Methoden, welche beim Design nicht-kovalenter Liganden angewandt wird. Unsere Arbeitsgruppe untersucht und in Folge wendet Prinzipien und fundamentale Konzepte an, welche relevant für das rationale Design und für die Optimierung von kovalenten Protein Liganden sind. Viele Untersuchungen zielen auf die Aufklärung von Inhibitionsmechanismen kovalenter Inhibitoren3,4 und die Rationalisierung bekannter Struktur-Aktivitäts-Beziehung ab. Zusätzlich, benchmarken wir die Genauigkeit vorhandener theoretischer Methoden zur Beschreibung der kovalenten Bindungsbildung.5
Das rationale Design kovalenter Inhibitoren von Grund auf ist praktisch unmöglich, weil Informationen über die geometrische Struktur des adressierten Enzyms essentiell sind. Zusätzlich sind theoretische Methoden zur Vorhersage von Enzym-Inhibitoren Komplexen meist sehr ungenau. Deshalb sollten Struktur-basierte Protokolle weniger fehleranfällig als Ansätze ohne strukturelle Informationen.
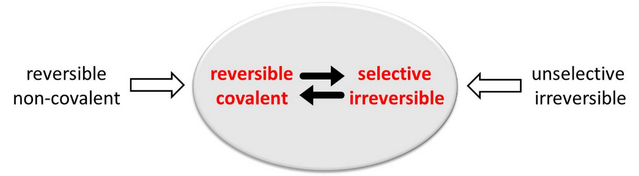
Abbildung 1: Mögliche Startstrukturen des Protokolls zum rationalen Design kovalenter Inhibitoren mit gewünschten Eigenschaften
Die Situation ist in Abbildung 1 dargestellt, in der mögliche Startpunkte zur Entwicklung von kovalenten Inhibitoren mit gewünschten Eigenschaften gezeigt werden. Um z.B. selektive irreversible Inhibitoren zu erhalten, kann man mit vorhandenen kristallographischen Daten von bekannten unselektiven irreversiblen Inhibitoren starten.6 Ein weiterer Startpunkt zur Entwicklung von kovalenten Inhibitoren sind kristallographische Daten von reversiblen nicht-kovalenten Inhibitoren. Das Prinzip wurde von anderen Gruppen angewandt, um kovalente Kinase Inhibitoren ausgehend von nicht-kovalenten zu entwickeln (Abbildung 2).
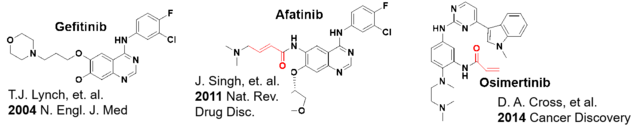
Abbildung 2: Entwicklung von kovalenten Kinase Inhibitoren ausgehend von nicht-kovalenten Verbindungen
Kovalenten Inhibitoren können in Verbindungen unterteilt werden, welche aktivierte Reste in der aktiven Tasche von Enzymen (orthosterische Inhibition) modulieren und Molekülen, welche in der Lage sind mit nicht-aktivierten Resten außerhalb der aktiven Tasche zu reagieren. Letztere können das Enzym inhibieren, da sie den Eingang der aktiven Tasche blockieren oder die Enzym Katalyse allosterisch deaktivieren. Im ersten Fall kann sich eine geringere Selektivität zeigen, weil die Region der aktiven Tasche konservierter ist.7,8 Im anderen Fall kann es komplizierter sein, weil man Einheiten hinzufügen muss, welche die Reste aktivieren. ASS scheint in diesem Fall ein Beispiel zu sein.9,10
Untersuchung von orthosterischen Inhibitoren
Est kürzlich konnten wir in Kollaboration mit Schirmeister aus Mainz ein Protokoll zum rationalen Design reversibler Inhibitoren ausgehend von irreversiblen entwickeln (Abbildung 3).11 Das Protokoll The protocol umfasst verschiedene computer-gestützte Schritte, welche nah an den dazugehörigen Experimenten verbunden sind, die die Vorhersagen realisieren und verifizieren. Als Startpunkt wurden die Untersuchungen an K11777 durchgeführt, ein allgemein bekannter irreversibler Inhibitor von Rhodesain. K11777 enhtält eine Vinylsulfon-Gruppe als elektrophilen Warhead, welcher irreversibel mit dem CYS25 Rest der aktiven Tasche von Rhodesain reagiert. Innerhalb dieser Untersuchungen haben wir zwei mögliche Modifikationen des Warheads entwickelt.11Während sich der erste als ungeeignet zeigt, 12 konnte durch Substitution eines Wasserstoffs der Doppelbindung durch ein Halogenatom die Thermodynamik der Reaktion in gewünschte Richtung geändert werden. In beiden Fällen zeigten sich die berechneten Vorhersagen als korret, das heißt der angewandte Ansatz scheint ausreichend genau zu sein. Das Protokoll umfasst Standard quantenchemische Ansätze um mögliche Warheads zu screenen. Hybrid-Ansätze, welche eine Kombination aus quantenmechanischen (QM) Methoden, die chemische Reaktionen beschreiben, und molekular mechanische (MM) Methoden, welche den Einfluss der Enzymumgebung beschreiben, wurden angewandt um die Inhibitionsreaktion innerhalb des Enzyms zu berechnen. Die Berechnungen starteten von kristallographischen Daten des kovalenten Enzym-Inhibitor Komplexes von K11777 und Rhodesain und der Pfad der Rückreaktion zum nicht-kovalenten Komplex wurden berechnet. Für weitere Informationen empfehlen wir einen Blick auf die Literatur11,12 In diesen Untersuchungen hat die Gruppe von Prof. Schirmeister (Mainz) jede Syntehese durchgeführt und sowohl in Lösung als auch im Enzym getestet. Zusätzlich haben sie bei den Docking Untersuchungen beigetragen. Weitere wichtige Beiträge kamen von der Gruppe Prof. Hellmich (Mainz), die die Enzym-Inhibitor Komplexe durch 19 F NMR Spektroskopie analysiert haben und Prof Tenzer (Mainz) , der durch Massenspektrometrie Messungen beigetragen hat, den Nachweis der kovalenten Bindungsbildung zu bringen.
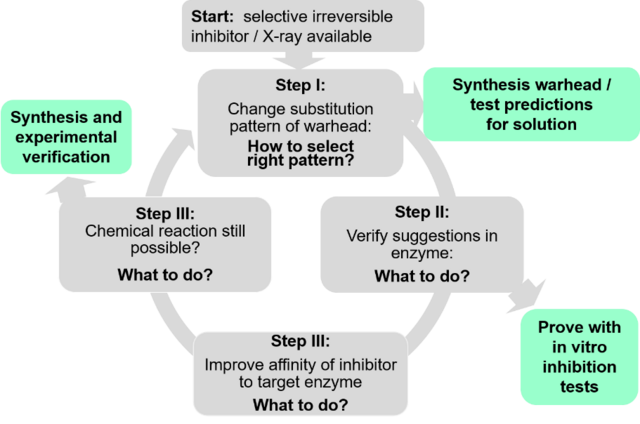
Abbildung 3: Protokoll zur Entwicklung reversibler kovalenter Inhibitoren unter Anwendung von bekannten irreversiblen als Startpunkt
Ebenfalls in enger Zusammenarbeit mit Prof. Schirmeister (Mainz) haben wir Struktur-Aktivitätsbeziehung (z.B. Regio- und Stereoselektivität) von Inhibitoren untersucht, deren Elektrophilie aufgrund von 3-gliedrigen Ringen oder Michael Systemen resulteiren.4,13,14 In einer derzeitigen Arbeit untersuchen wir Inhibitionsmechanismen von Verbindungen, welche Nitro-Alkene als Warheads tragen. Diese sind wesentlich potenten als Vinylsulfon-basierte Inhibitoren und scheinen verschiedene Inhibitonsmechanismen zu haben.,15 Derzeit werden auch Chloro- oder NItril-substituierte 1,4-Naphtoquinone als Warheads in Kooperation mit Prof. Opatz (Mainz) and Prof. Schirmeister (Mainz) untersucht, welche in Form einer Michael-Addition und/oder Substitution reagieren.
|
|
Abbildung 4: links: Berechneter nicht-kovalenter Komplex von Carbamat-basiertem Warhead und Cathepsin B. rechts: Computed non-covalent complex of the carbamate-based warhead and cathepsin B. right hand side: Potentialhyperfläche des Zwischenschrittes | |
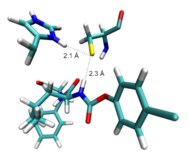
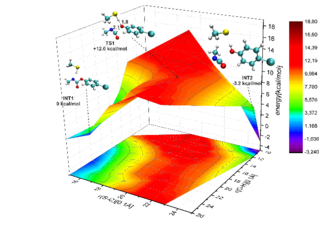
In Zusammenarbeit mit Prof. Gütschow (Bonn) haben wir die Inhibitionsreaktion von Carbamat-basierten Komponenten in Cathepsin B untersucht. Der zugrundeliegende Mechanismus scheint komplizierter zu sein als oben diskutierte Additionen an aktivierten Doppelbindungen, weil die Reaktion aus mehreren Schritten besteht, welche häufig den Prozess des Protonontransfers beinhaltet. In derzeitigen Untersuchungen vergleichen wir verschiedene Reaktionswege, um diesen komplizierten Sachverhalt zu erklären.
|
|
Abbildung 5: K11777 (links) und sein Derivat 7a (rechts) | |
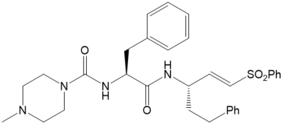
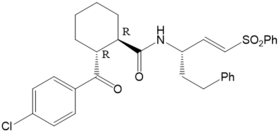
Für K11777 und seinem Derivat 7a (Abbildung 5) haben wir Untersuchungen durchgeführt, ob gemessene Trends der kinact Werte zur Inhibition von Rhodesain16 mittels der Theorie erklärt werden kann (kinact (K11777) = 0.029 s-1; kinact (7a) = 0.00051 s-1). Die berechneten Reaktionsprofile der kovalenten Bindungsbildung zeigten keine Unterschiede wie im Fall der Untersuchung der Stereoselektivität von der Inhibition des Cathepsin B durch E64C verwandte Strukturen.13,17 Im Gegensatz dazu zeigen MD Simulationen, dass die Unterschiede aufgrund der geometrischen Orientierung des nicht-kovaleten Komplexes liegen. Diese sagen für K11777 eine deutlich geringere Distanz zwischen elektrophilem Zentrum des Warheads und Cys25 aus als für 7a, bei dem größere Distanzen gefunden wurden. In Kooperation mit Prof. Hellmich (Jena), Prof. Schindelin (Würzburg), aundProf. Ochsenfeld (LMU Munich) haben wir den Bindungsmechanismus von kovalenten Inhibitoren von Tryparedoxin (Tpx) von T. brucei untersucht.
(1) Singh, J.; Petter, R. C.; Baillie, T. A.; Whitty, A. The resurgence of covalent drugs. Nature reviews Drug discovery 2011, 10, 307-317.
(2) Ostrem, J. M.; Peters, U.; Sos, M. L.; Wells, J. A.; Shokat, K. M. K-Ras (G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548-551.
(3) Paasche, A.; Zipper, A.; Schäfer, S.; Ziebuhr, J.; Schirmeister, T.; Engels, B. Evidence for substrate binding-induced zwitterion formation in the catalytic Cys-His dyad of the SARS-CoV main protease. Biochemistry 2014, 53, 5930-5946.
(4) Mladenovic, M.; Schirmeister, T.; Thiel, S.; Thiel, W.; Engels, B. The importance of the active site histidine for the activity of epoxide- or aziridine-based inhibitors of cysteine proteases. Chemmedchem 2007, 2, 120-128.
(5) Paasche, A.; Schirmeister, T.; Engels, B. Benchmark Study for the Cysteine-Histidine Proton Transfer Reaction in a Protein Environment: Gas Phase, COSMO, QM/MM Approaches. Journal of Chemical Theory and Computation 2013, 9, 1765-1777.
(6) Schmidt, T. C.; Welker, A.; Rieger, M.; Sahu, P. K.; Sotriffer, C. A.; Schirmeister, T.; Engels, B. Protocol for Rational Design of Covalently Interacting Inhibitors. Chemphyschem 2014, 15, 3226-3235.
(7) Nussinov, R.; Tsai, C.-J. The design of covalent allosteric drugs. Annual review of pharmacology and toxicology 2015, 55, 249-267.
(8) Wu, H. M.; Bock, S.; Snitko, M.; Berger, T.; Weidner, T.; Holloway, S.; Kanitz, M.; Diederich, W. E.; Steuber, H.; Walter, C.; Hofmann, D.; Weissbrich, B.; Spannaus, R.; Acosta, E. G.; Bartenschlager, R.; Engels, B.; Schirmeister, T.; Bodem, J. Novel Dengue Virus NS2B/NS3 Protease Inhibitors. Antimicrobial Agents and Chemotherapy 2015, 59, 1100-1109.
(9) Toth, L.; Muszbek, L.; Komaromi, I. Mechanism of the irreversible inhibition of human cyclooxygenase-1 by aspirin as predicted by QM/MM calculations. Journal of Molecular Graphics & Modelling 2013, 40, 99-109.
(10) Tosco, P.; Lazzarato, L. Mechanistic Insights into Cyclooxygenase Irreversible Inactivation by Aspirin. Chemmedchem 2009, 4, 939-945.
(11) Schirmeister, T.; Kesselring, J.; Jung, S.; Schneider, T. H.; Weickert, A.; Becker, J.; Lee, W.; Bamberger, D.; Wich, P. R.; Distler, U.; Tenzer, S.; Johe, P.; Hellmich, U. A.; Engels, B. Quantum Chemical-Based Protocol for the Rational Design of Covalent Inhibitors. Journal of the American Chemical Society 2016, 138, 8332-8335.
(12) Schneider, T. H.; Rieger, M.; Ansorg, K.; Sobolev, A. N.; Schirmeister, T.; Engels, B.; Grabowsky, S. Vinyl sulfone building blocks in covalently reversible reactions with thiols. New Journal of Chemistry 2015, 39, 5841-5853.
(13) Mladenovic, M.; Junold, K.; Fink, R. F.; Thiel, W.; Schirmeister, T.; Engels, B. Atomistic insights into the inhibition of cysteine proteases: First QM/MM calculations clarifying the regiospecificity and the inhibition potency of epoxide- and aziridine-based inhibitors. Journal of Physical Chemistry B 2008, 112, 5458-5469.
(14) Buback, V.; Mladenovic, M.; Engels, B.; Schirmeister, T. Rational Design of Improved Aziridine-Based Inhibitors of Cysteine Proteases. Journal of Physical Chemistry B 2009, 113, 5282-5289.
(15) Latorre, A.; Schirmeister, T.; Kesselring, J.; Jung, S.; Johe, P.; Hellmich, U. A.; Heilos, A.; Engels, B.; Krauth-Siegel, R. L.; Dirdjaja, N.; Bou-Iserte, L.; Rodriguez, S.; Gonzalez, F. V. Dipeptidyl Nitroalkenes as Potent Reversible Inhibitors of Cysteine Proteases Rhodesain and Cruzain. Acs Medicinal Chemistry Letters 2016, 7, 1073-1076.
(16) Kerr, I. D.; Lee, J. H.; Farady, C. J.; Marion, R.; Rickert, M.; Sajid, M.; Pandey, K. C.; Caffrey, C. R.; Legac, J.; Hansell, E. Vinyl sulfones as antiparasitic agents and a structural basis for drug design. Journal of Biological Chemistry 2009, 284, 25697-25703.
(17) Mladenovic, M.; Fink, R. F.; Thiel, W.; Schirmeister, T.; Engels, B. On the origin of the stabilization of the zwitterionic resting state of cysteine proteases: A theoretical study. Journal of the American Chemical Society 2008, 130, 8696-8705.