Medicinal/ Biological Chemistry
Investigation of inhibition mechanisms covalent inhibitors
Due to their various advantages, covalent ligands currently experience an intensive renaissance not only in academic but also in industrial developments.1 Regarding their exploitation as drugs, they are more efficient in vivo and can be administered in lower doses and with reduced frequency due to their prolonged residence time at the target. Moreover, the concept of covalently modifying a protein offers the possibility to address targets that were assumed completely intractable for small-molecular drugs because specific binding pockets are absent (“drug the undruggable”).2 So far, however, most covalent ligands have been found serendipitously not the least because the tools for their rational optimization or de novo design are not as well established and elaborated as the corresponding methods for the design of non-covalent ligands. Our workgroup elucidates and subsequent apply principles and fundamental concepts relevant for the rational design and optimization of covalent protein ligands. Several investigations are also devoted to the clarification of inhibition mechanisms of covalent inhibitors3,4 and the rationalization of known structure-activities relationships. Additionally, we benchmark the accuracy of available theoretical approaches for the description of the covalent bond forming step.5
The rational design of covalent inhibitors from scratch is nearly impossible because some information about the geometrical structure of the target enzyme are essential. Furthermore, theoretical approaches to predict enzyme-inhibitor complexes are often very inaccurate. Hence, structure-based protocols should be less error prone than approaches without any structural information.
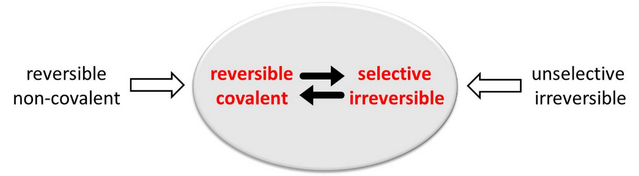
Figure 1: Possible starting structure for protocols for a rational design of covalent inhibitors with desired properties
The situation is sketched in Figure 1, which gives possible starting points to develop covalent inhibitors with desired properties. To obtain e.g. selective irreversible one could start from available crystallographic data of known unselective irreversible inhibitors.6 Another starting point to develop covalent inhibitors are crystallographic data of reversible non-covalent inhibitors. This principle was used by other groups to develop covalent kinase inhibitors starting from non-covalent ones (Figure 2).
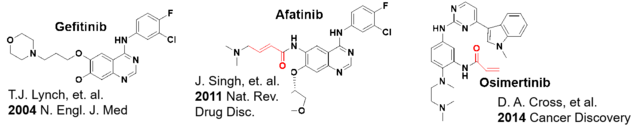
Figure 2: Development of covalent kinase inhibitors starting from non-covalent compounds
Covalent inhibitors can be divided into compounds, which modulate activated residues in the active site, of enzymes (orthosteric inhibition) and molecules, which are able to react with non-activated residues sitting outside of the active site. The latter can inhibit the enzyme because they block the entrance to the active site or because they disable the enzyme catalysis allosterically. The former may show less selectivity because the active site region is more conserved.7,8 The structure of the latter may be more complicated because they may have to include units, which activate the target residue. ASS seems to be such an example.9,10
Investigations of orthosteric inhibitors
Recently, in collaboration with the Schirmeister in Mainz we developed a protocol for the rational design of reversible inhibitors starting from known irreversible ones (Figure 3).11 The protocol comprises various computational steps, which are closely interconnected with the corresponding experiments, which realize and verify the predictions. The investigations used K11777, a well-known irreversible inhibitor of rhodesain, as starting point. K11777 possesses a vinylsulfone group as electrophilic warhead, which reacts irreversibly with the CYS25 residue of the active site of rhodesain. Within the investigations, we developed two possible modification of the warhead.11 While the first one turned out to be inappropriate12 the idea to substitute one hydrogen of the double bond by a halogen atom changed the thermodynamic of the reaction in the desired way. In both cases, the computed predictions turned out to be correct, i.e. the used approaches seem to be sufficiently accurate. The protocol comprises standard quantum chemical approaches to screen possible warheads. Hybrid approaches, which combine quantum mechanical (QM) methods to describe the chemical reactions with molecular mechanical (MM) methods to fold in the influence of the enzyme environment, were employed to compute the inhibition reactions within the enzyme. The computations started from the crystallographic data for the covalent enzyme-inhibitor complex of K11777 and rhodesain and computed the reaction path backwards to the non-covalent complex. For more information, we refer to the literature.11,12 In these investigations the group of Prof. Schirmeister (Mainz) performed all synthesis and testing in enzyme as well as in solution. Additionally, they contributed docking investigations. Further important contributions came from the group of Prof. Hellmich (Mainz) who analyzed the enzyme-inhibitor complexes by 19F and Prof Tenzer (Mainz) who contributed mass spectrometry measurements to proof that a covalent bond is formed.
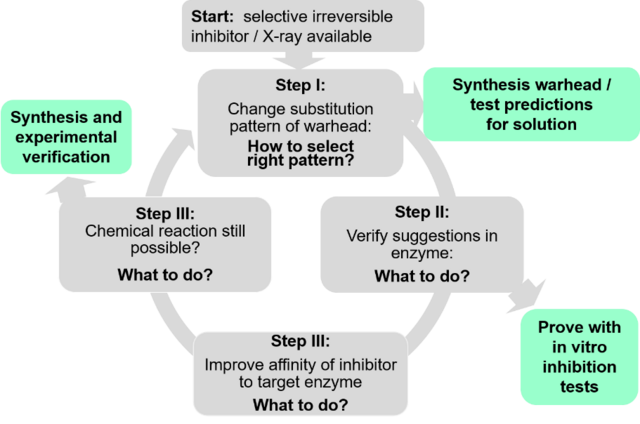
Figure 3: Protocol to develop reversible covalent inhibitors using known irreversible ones as starting point.
Additionally, in close collaboration of Prof. Schirmeister (Mainz) we investigated structure-activity relationships (e.g. regio and stereoselectivity) of inhibitors whose electrophilicity results from three-membered rings or various Michael systems.4,13,14 In ongoing work, we investigate the inhibition mechanism of compounds with nitro-alkenes as warheads. They are considerably more potent than vinylsulfone-based inhibitors and seem to possess a different inhibition mechanism.15 In ongoing investigations we also investigate chloro- or nitrile-substituted 1,4-napthoquinones as warheads which can undergo Michael-type addition and/or substitution reactions in cooperation with Prof. Opatz (Mainz) and Prof. Schirmeister (Mainz).
|
|
Figure 4: left hand side: Computed non-covalent complex of the carbamate-based warhead and cathepsin B. right hand side: Potential energy surface of an intermediate step of the corresponding | |
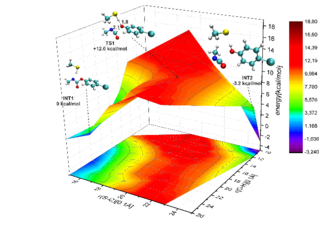
In cooperation with Prof. Gütschow (Bonn) we investigate the inhibition reaction of carbamate-based compounds with cathepsin B. The underlying mechanism seem to be more complicated than the above-discussed additions to activated double bonds because it consists of several steps which often involves proton transfer processes. In an ongoing study, we compare different reaction routes to shed some light on this complicated issue.
|
|
Figure 5: K11777 (left hand side) and its derivative 7a (right hand side) | |
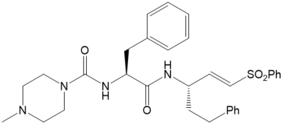
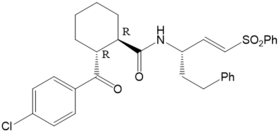
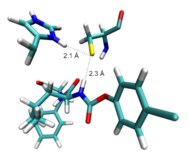
For K11777 and its Derivative 7a (Figure 5) we investigated if the measured trends in the kinact values for the inhibition of rhodesain16 can be explained by theory (kinact (K11777) = 0.029 s-1; kinact (7a) = 0.00051 s-1). The computed reaction profiles of the covalent bond formation do not show differences as already found within the investigation of the stereoselectivity of the inhibition cathepsin B by E64C related compounds.13,17 In contrast, MD simulation indicate that the differences arise from the geometrical orientation of the non-covalent complex. They predict that for K11777 the distance between the electrophilic center of the warhead and Cys25 is considerably smaller than for 7a, for which larger distances are found. In cooperation with Prof. Hellmich (Mainz), Prof. Schindelin (Würzburg), and Prof. Ochsenfeld (LMU Munich) we investigate the binding mechanisms of covalent inhibitors of Tryparedoxin (Tpx) from T. brucei.
(1) Singh, J.; Petter, R. C.; Baillie, T. A.; Whitty, A. The resurgence of covalent drugs. Nature reviews Drug discovery 2011, 10, 307-317.
(2) Ostrem, J. M.; Peters, U.; Sos, M. L.; Wells, J. A.; Shokat, K. M. K-Ras (G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548-551.
(3) Paasche, A.; Zipper, A.; Schäfer, S.; Ziebuhr, J.; Schirmeister, T.; Engels, B. Evidence for substrate binding-induced zwitterion formation in the catalytic Cys-His dyad of the SARS-CoV main protease. Biochemistry 2014, 53, 5930-5946.
(4) Mladenovic, M.; Schirmeister, T.; Thiel, S.; Thiel, W.; Engels, B. The importance of the active site histidine for the activity of epoxide- or aziridine-based inhibitors of cysteine proteases. Chemmedchem 2007, 2, 120-128.
(5) Paasche, A.; Schirmeister, T.; Engels, B. Benchmark Study for the Cysteine-Histidine Proton Transfer Reaction in a Protein Environment: Gas Phase, COSMO, QM/MM Approaches. Journal of Chemical Theory and Computation 2013, 9, 1765-1777.
(6) Schmidt, T. C.; Welker, A.; Rieger, M.; Sahu, P. K.; Sotriffer, C. A.; Schirmeister, T.; Engels, B. Protocol for Rational Design of Covalently Interacting Inhibitors. Chemphyschem 2014, 15, 3226-3235.
(7) Nussinov, R.; Tsai, C.-J. The design of covalent allosteric drugs. Annual review of pharmacology and toxicology 2015, 55, 249-267.
(8) Wu, H. M.; Bock, S.; Snitko, M.; Berger, T.; Weidner, T.; Holloway, S.; Kanitz, M.; Diederich, W. E.; Steuber, H.; Walter, C.; Hofmann, D.; Weissbrich, B.; Spannaus, R.; Acosta, E. G.; Bartenschlager, R.; Engels, B.; Schirmeister, T.; Bodem, J. Novel Dengue Virus NS2B/NS3 Protease Inhibitors. Antimicrobial Agents and Chemotherapy 2015, 59, 1100-1109.
(9) Toth, L.; Muszbek, L.; Komaromi, I. Mechanism of the irreversible inhibition of human cyclooxygenase-1 by aspirin as predicted by QM/MM calculations. Journal of Molecular Graphics & Modelling 2013, 40, 99-109.
(10) Tosco, P.; Lazzarato, L. Mechanistic Insights into Cyclooxygenase Irreversible Inactivation by Aspirin. Chemmedchem 2009, 4, 939-945.
(11) Schirmeister, T.; Kesselring, J.; Jung, S.; Schneider, T. H.; Weickert, A.; Becker, J.; Lee, W.; Bamberger, D.; Wich, P. R.; Distler, U.; Tenzer, S.; Johe, P.; Hellmich, U. A.; Engels, B. Quantum Chemical-Based Protocol for the Rational Design of Covalent Inhibitors. Journal of the American Chemical Society 2016, 138, 8332-8335.
(12) Schneider, T. H.; Rieger, M.; Ansorg, K.; Sobolev, A. N.; Schirmeister, T.; Engels, B.; Grabowsky, S. Vinyl sulfone building blocks in covalently reversible reactions with thiols. New Journal of Chemistry 2015, 39, 5841-5853.
(13) Mladenovic, M.; Junold, K.; Fink, R. F.; Thiel, W.; Schirmeister, T.; Engels, B. Atomistic insights into the inhibition of cysteine proteases: First QM/MM calculations clarifying the regiospecificity and the inhibition potency of epoxide- and aziridine-based inhibitors. Journal of Physical Chemistry B 2008, 112, 5458-5469.
(14) Buback, V.; Mladenovic, M.; Engels, B.; Schirmeister, T. Rational Design of Improved Aziridine-Based Inhibitors of Cysteine Proteases. Journal of Physical Chemistry B 2009, 113, 5282-5289.
(15) Latorre, A.; Schirmeister, T.; Kesselring, J.; Jung, S.; Johe, P.; Hellmich, U. A.; Heilos, A.; Engels, B.; Krauth-Siegel, R. L.; Dirdjaja, N.; Bou-Iserte, L.; Rodriguez, S.; Gonzalez, F. V. Dipeptidyl Nitroalkenes as Potent Reversible Inhibitors of Cysteine Proteases Rhodesain and Cruzain. Acs Medicinal Chemistry Letters 2016, 7, 1073-1076.
(16) Kerr, I. D.; Lee, J. H.; Farady, C. J.; Marion, R.; Rickert, M.; Sajid, M.; Pandey, K. C.; Caffrey, C. R.; Legac, J.; Hansell, E. Vinyl sulfones as antiparasitic agents and a structural basis for drug design. Journal of Biological Chemistry 2009, 284, 25697-25703.
(17) Mladenovic, M.; Fink, R. F.; Thiel, W.; Schirmeister, T.; Engels, B. On the origin of the stabilization of the zwitterionic resting state of cysteine proteases: A theoretical study. Journal of the American Chemical Society 2008, 130, 8696-8705.