Recent Publications
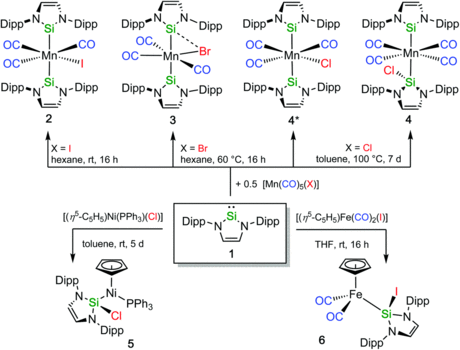
N-Heterocyclic silylene complexes of nickel(0)
E. Glok, L. Werner, U. Radius, Dalton. Trans. 2025, 54, 14846-14861.
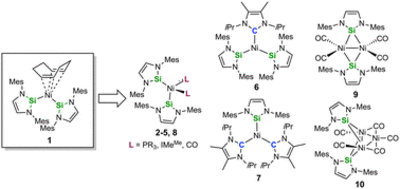
Reactivity studies on the nickel complex [Ni(Mes2NHSi)2(COD)] (1), ligated with the N-heterocyclic silylene 1,3-bis(2,6-dimesityl)-1,3-diaza-2-silacyclopent-4-en-2-ylidene (= Mes2NHSi; COD = 1,5-cyclooctadiene) are presented. Treatment of 1 with two equivalents of the phosphines PMe3, PEt3 and PnBu3, and the N-heterocyclic carbene 1,3,4,5-tetramethyl-imidazolin-2-ylidene (IMeMe) led to substitution of the COD (and not the NHSi) ligand to yield the heteroleptic substituted complexes [Ni(Mes2NHSi)2(L)2] (L = PMe3 (2), PEt3 (3), PnBu3 (4), IMeMe (5)) in good yields. The reaction of [Ni(Mes2NHSi)2(COD)] with IiPrMe (1,3-diisopropyl-4,5-dimethyl-imidazolin-2-ylidene) and the reaction of [Ni(IiPrMe)2(η2-COE)] (COE = cyclooctene) with Mes2NHSi afforded mixtures of heteroleptic three-coordinated nickel complexes [Ni(Mes2NHSi)2(IiPrMe)] (6) and [Ni(Mes2NHSi)(IiPrMe)2] (7). Complex 1 reacts with carbon monoxide also under substitution of the COD ligand to afforded mixtures of mononuclear [Ni(Mes2NHSi)2(CO)2] (8) and dinuclear, silylene-bridged [{Ni(μ-Mes2NHSi)(CO)2}2] (9). Treatment of [Ni(CO)4] with Mes2NHSi also led to formation of the complexes 8 and 9, and, in addition, to the trinuclear Chini-type cluster complex [Ni3(μ-Mes2NHSi)2(μ-CO)2(CO)3] (10). In contrast to known related complexes of NHCs, complex 1 does not react with aryl fluorides and aryl chlorides and decomposes in the reaction with organyl bromides and iodides.
Radical anions of 1,1-azoliumdithiocarboxylates
M. S. Luff, K. Oppel, I. Krummenacher, H. Braunschweig, U. Radius, Chem. Commun. 2024, 60, 14447-14450.
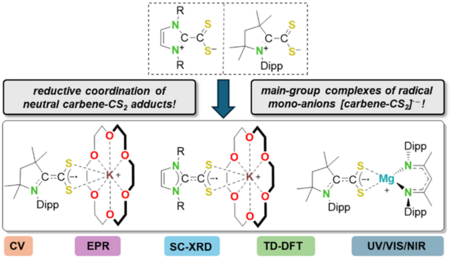
The 1,1-azoliumdithiocarboxylate cAACMe-CS2 (1a) was prepared and its redox chemistry was evaluated and compared to NHC-based dithiocarboxylates IDipp‑CS2 (1b) and IMes-CS2 (1c). Radical anions [carbene‑CS2]·– were prepared by metallic reduction as the potassium or magnesium ion complexes [K(18‑crown‑6)(cAACMe-CS2)] (2a), [K(18‑crown‑6)(NHC-CS2)] (NHC = IDipp: 2b, IMes: 2c), and [Mg(DippNacNac)(cAACMe-CS2)] (3) and extensively characterized (SC-XRD, EPR, UV/VIS/NIR, DFT). These complexes represent the first examples of isolated radical anions of 1,1‑dithiolenes.
NHC Aluminum Chemistry on the Rise
L. Werner, U. Radius, Dalton. Trans. 2024, 53, 16436-16454.
This perspective highlights recent developments of the use of N-heterocyclic carbenes (NHCs) and cyclic (alkyl)(amino)carbenes (cAACs) in alane and aluminum organyl chemistry. Especially in the last few years this flourishing research field led to some remarkable discoveries including various substitution patterns at the central aluminum atom, different oxidation states, neutral and charged compounds with varying coordination numbers and unique reactivities. Thereby NHCs play a vital role in the stabilization of these otherwise highly reactive compounds, which would not be realizable without the use of this intriguing class of ligands. Nevertheless, main group hydrides and especially NHC ligated alanes also tend to undergo NHC decomposition reactions, which are part of ongoing research and provide important information for NHC research in general.
Construction of a Boron Chain on a Single Metal by Dehydrocoupling of Borane
C. Luz, K. Oppel, L. Endres, R. D. Dewhurst, H. Braunschweig, U. Radius, J. Am. Chem. Soc. 2024, 146, 23741-23751.
Borane coordination, B–H borane bond activation, and borane catenation via metal-mediated dehydrocoupling to form electron-precise B–B bonds is reported. The reaction of trans-[M(IMes)2Cl4] (M = W, Mo) (IMes = 1,3-bis(2,4,6-trimethylphenyl)imidazolin-2-ylidene) with the borates Li[BH3R] (R = Mes, Dur; Mes = 2,4,6-Me3C6H2, Dur = 2,3,5,6-Me4C6H) afforded the complexes [M(IMes)(η2-H2BR)2(η1-H2BR)] (M = W: R = Mes 1, R = Dur 3; M = Mo: R = Mes 2, R = Dur 4). Three borane ligands are coordinated in 1 – 4 to the group 6 metal atom via five (σ-B–H) bonds. Reaction of 1 with the phosphines PMe3 and PEt3, respectively, led to elimination of one of the borane ligands and afforded the hydrido (σ-B–H)-boryl bis-(σ-B–H)-borane complexes trans-[W(IMes)(PR3)(η1-HBMes)(η2-H2BMes)(H)] (R = Me, 5, R = Et 6), in which the metal atom inserted into one of the remaining σ-B–H bonds of the borane ligands. Reaction of 1 with an additional equivalent borane BH2Mes resulted in borane dehydrocoupling and formation of the complex [W(IMes)(η4-BH2Mes-BMes-BMes-BH2Mes)] 7, featuring a unique B4 chain as ligand. Reaction of trans-[W(IMes)2Cl4] with NaBH4 also led to B–B coupling, and the metallaborane cluster [{W(IMes)(BH4)}2(B5H9)] 9 was formed, in which two tungsten atoms bridge a B5 chain.

The Tetracyanoborate Anion as Building Block for the Heterocubane Cluster Cage [Cr4{B(CN)4}4]4–
M. S. Luff, R. Bertermann, M. Finze, U. Radius, Chem. Commun. 2024, 60, 8621-8624.
The tetracyanoborate anion [B(CN)4]– (TCB) was utilized as a building block for the synthesis of polynuclear chromium carbonyl compounds upon photolytic reaction with [Cr(CO)6]. Up to four kN-coordinated cyano groups of TCB can be involved in binding to chromium, giving mixtures of [B(CN)4–x{CN–Cr(CO)5}x]- (x = 1–4; 1–4) and [{Cr(CO)4(B(CN)4)}2]2– (5). The reaction of [B(CN)4]– with fac-[Cr(CO)3(MeCN)3] allowed for the isolation and characterization of salts of the tetraanionic heterocubane cage [{Cr(CO)3(B(CN)4)}4]4–.
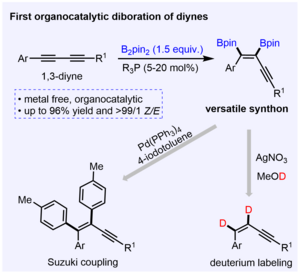
Phosphine-catalyzed 1,2-cis-diboration of 1,3-butadiynes
W. Li, R. Ricker, K. L. Chan, P. F. Lau, N. W. Buchbinder, J. Krebs, A. Friedrich, Z. Lin, W. L. Santos, U. Radius, T. B. Marder, Chem. Eur. J. 2024, 30, e202401235.
Trialkyl phosphines PMe3 and PEt3 catalyze the 1,2-cis-diboration of 1,3-butadiynes to give 1,2-diboryl enynes. The products were utilized to synthesize 1,1,2,4-tetraaryl enynes using a Suzuki-Miyaura protocol and can readily undergo proto-deborylation.
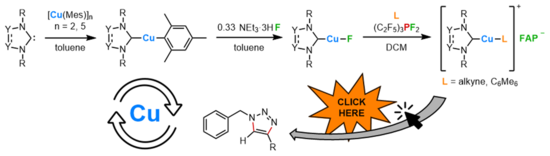
M. Riethmann, S. A. Föhrenbacher, H. Keiling, N. V. Ignat’ev, M. Finze, U. Radius, Inorg. Chem. 2024, 63, 8351-8365.
We herein report the convenient synthesis of different N-heterocyclic carbene (NHC)- and cyclic (alkyl)(amino)carbene (cAAC) ligated copper cations using the weakly coordinating tris(pentafluoroethyl)trifluorophosphate counterion (FAP anion, [(C2F5)3PF3]−). The reaction of the fluorido complexes [(carbene)CuF] (carbene = NHC, cAACMe) 2a–f and the tris(pentafluoroethyl)difluorophosphorane (C2F5)3PF2 in the presence of alkynes or arenes led to fluoride transfer from Cu to the phosphorane with formation of the cationic transition metal complexes [(carbene)Cu(L)]+ and the weakly coordinating counter-anion [(C2F5)3PF3]− (FAP). Using this method, the complexes [(IDipp)Cu(L)]+FAP- (IDipp = 1,3-bis(2,6-di-iso-propylphenyl)-imidazolin-2-ylidene; L = PhC≡CPh, 4d; PhC≡CMe, 5d), [(cAACMe)Cu(L)]+FAP- (cAACMe = 1-(2,6-di-iso-propylphenyl)-3,3,5,5-tetramethyl-pyrrolidin-2-ylidene; L = PhC≡CPh, 4f; PhC≡CMe, 5f), [(SIDipp)Cu(C6Me6)]+FAP- (6e), (SIDipp = 1,3-bis(2,6-di-iso-propylphenyl)-imidazolidine-2-ylidene), and [(cAACMe)Cu(C6Me6)]+FAP- (6f) have been synthesized and characterized. The complexes [(IDipp)Cu(C6Me6)]+FAP- (6d), and [(cAACMe)Cu(C6Me6)]+FAP- (6f) have been used as catalysts for the copper(I) catalyzed cycloaddition of benzyl azide to terminal alkynes.
Y. P. Budiman, R. N. Perutz, P. Steel, U. Radius, T. B. Marder, Chem. Rev. 2024, 124, 4822-4862.
The synthesis of organic compounds efficiently via fewer steps but in higher yields is desirable as this reduces energy and reagent use, waste production, and thus environmental impact as well as cost. The reactivity of C–H bonds ortho-to-fluorine substituents in (poly)fluoroarenes with metal centers is enhanced relative to meta and para positions. Thus, direct C–H functionalization of (poly)fluoroarenes without prefunctionalization is becoming a significant area of research in organic chemistry. Novel and selective methodologies to functionalize (poly)fluorinated arenes by taking advantage of the reactivity of C–H bonds ortho-to-C–F bonds are continuously being developed. This review summarizes the reasons for the enhanced reactivity and the consequent developments in the synthesis of valuable (poly)fluoroarene-containing organic compounds.

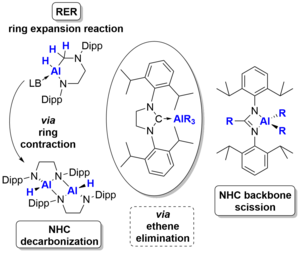
L. Werner, U. Radius, Angew. Chem. Int. Ed. 2024, 63, e202403639.
The reaction of the amine-stabilized alane (NMe3)∙AlH3 1 with the backbone-saturated N-heterocyclic carbene (NHC) SIDipp (SIDipp = 1,3-bis-{2,6-di-iso-propyl-phenyl}-imidazolidin-2-ylidene) at 0°C yielded the NHC alane adduct (SIDipp)∙AlH3 2. Reaction at elevated temperatures or prolonged reaction at room temperature gave the product of a ring expansion reaction (RER) of the NHC, 3∙(NMe3). Subsequent reaction of the latter with sterically less hindered NHCs (IMeMe {= 1,3,4,5-tetramethyl-imidazolin-2-ylidene}, IiPrMe {= 1,3-di-iso-propyl-4,5-dimethyl-imidazolin-2-ylidene}, and IiPr {= 1,3-di-iso-propyl-imidazolin-2-ylidene}) afforded the NHC-stabilized RER-products (NHC)∙AlH(RER-SIDippH2) 3∙(NHC) (NHC = IMeMe, IiPrMe, IiPr), while no reaction was observed with the sterically more demanding NHCs IDipp (= 1,3-bis-{2,6-di-iso-propyl-phenyl}-imidazolin-2-ylidene), SIDipp and ItBu (= 1,3-di-tert-butyl-imidazolin-2-ylidene). The compounds 3∙(NHC) were also obtained starting from (SIDipp)∙AlH3 2 and NHC at room temperature. Heating solutions of (SIDipp)∙AlH3 2 without additional base to 95°C resulted in decarbonization of the NHC and substitution of the carbene carbon atom with aluminum hydride under loss of ethene. Subsequent dimerization afforded cis-[AlH{-N(Dipp)CH2CH2N(Dipp)}]2 4_dimer. Heating solutions of the NHC-ligated aluminum alkyls (SIDipp)∙AlR3 2R (R = Me, Et) to 145°C instead led to complete scission of the NHC backbone with evolution of ethene and isolation of the dialkylaluminium(III) amidinates {DippNC(R)NDipp}AlR2 5R (R = Me, Et).
L. Werner, J. Hagn, A. Gerstner, U. Radius, Dalton. Trans. 2024, 53, 5932-5946.
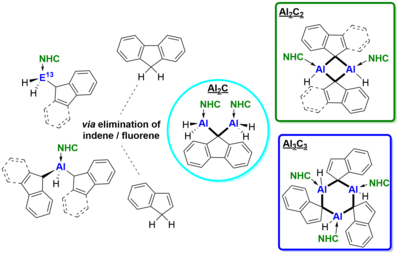
Indenyl-(Ind) and fluorenyl-(Fl) substituted NHC-stabilized alanes and gallanes (NHC)·EH2R 1-12 (NHC = IiPrMe, IiPr, IMeMe; E = Al, Ga; R = Ind, Fl) were prepared via reaction of the corresponding NHC-iodoalanes and -gallanes with LiInd and LiFl respectively. Analogously, the alane adducts with two Ind/Fl substituents (NHC)·AlHR2 13-18 (NHC = IiPrMe, IiPr, IMeMe; R = Ind, Fl) were obtained by using two equivalents of LiInd/LiFl. Elimination of indene and fluorene was induced thermally affording unusual dimeric and trimeric NHC-alane adducts {(NHC)·AlH2}2-m-Fl 19-20 and {(NHC)·AlH-m-R}n 21-23 (R = Ind, Fl; n = 2, 3) with bridging indenyl and fluorenyl ligands.
Pd-Catalyzed Oxidative C–H Arylation of (Poly)fluoroarenes with Aryl Pinacol Boronates
Y. P. Budiman, M. H. Putra, M. R. Ramadhan, C. Luz, I. Z. Ghafara, R. Rustaman, E. E. Ernawati, T. Mayanti, A. Groß, U. Radius, T. B. Marder, Chem. As. J. 2024, 19, e202400094.
We report the synergistic combination of Pd(OAc)2 and Ag2O for the oxidative C–H arylation of (poly)fluoroarenes with aryl pinacol boronates (Ar-Bpin) in DMF as the solvent. This procedure can be conducted easily in air, and without using additional ligands, to afford the fluorinated unsymmetrical biaryl products in up to 98% yield. Experimental studies suggest that the formation of [PdL2(C6F5)2] in DMF as coordinating solvent does not take place under the reaction conditions as it is stable to reductive elimination and thus would deactivate the catalyst. Thus, the intermediate [Pd(DMF)2(ArF)(Ar)] must be formed selectively to give desired arylation products. DFT calculations predict a low barrier (5.87 kcal/mol) for the concerted metalation deprotonation (CMD) process between C6F5H and the Pd(II) species formed after transmetalation between the Pd(II)X2 complex and aryl-Bpin forms a Pd-Arrich species; thus a Pd(Arrich)(Arpoor) complex is generated selectively which undergoes reductive elimination to generate the unsymmetrical biaryl product.

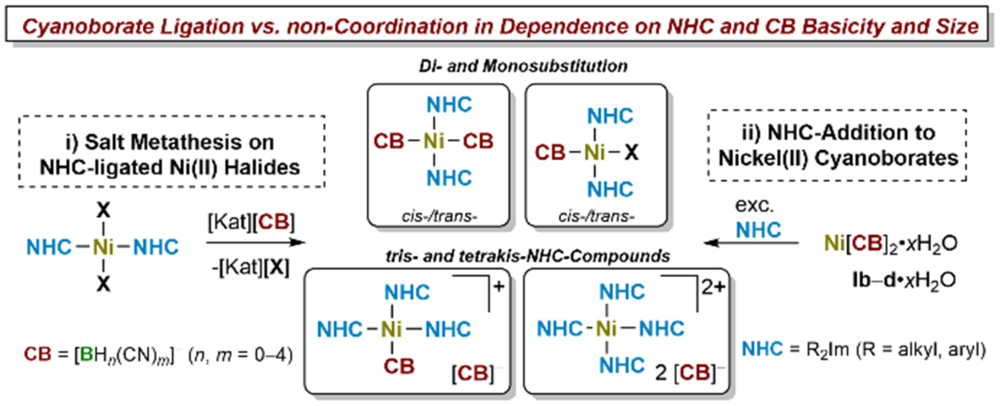
NHC-ligated Nickel(II) Cyanoborate Complexes and Salts
M. S. Luff, L. Walther, M. Finze, U. Radius, Dalton Trans. 2024, 53, 5391-5400.
A comprehensive study on the synthesis and characterization of NHC-ligated nickel(II) cyanoborates (CBs) is presented (NHC = N-heterocyclic carbene). Nickel(II) cyanoborates Ni[BH2(CN)2]2·H2O (Ib·H2O), Ni[BH(CN)3]2·0.5H2O (Ic·0.5H2O), Ni[B(CN)4]2·H2O (Id·H2O) were reacted with selected NHCs of different steric size. The reaction of the nickel cyanoborates with small to medium-sized NHCs Me2ImMe and iPr2Im (R2Im = 1,3-di-organyl-imidazolin-2-ylidene; R2ImMe = 1,3-di-organyl-4,5-dimethyl-imidazolin-2-ylidene) afforded cyanoborate salts containing the rare homoleptic fourfold NHC-ligated nickel(II) cations [Ni(NHC)4]2+ (NHC = Me2ImMe (1c–d), iPr2Im (2c–d)) and cyanoborate counter-anions. Bulkier NHCs such as Mes2Im and Dipp2Im afforded complexes trans-[Ni(NHC)2(CB)2] (trans-4b, trans-5c). For the combination of the cyanoborate anion [BH2(CN)2]– and iPr2ImMe the salt of the tris-NHC complex [Ni(iPr2ImMe)3(NC‑BH2CN)][BH2(CN)2] (3b) was isolated. Salt metathesis of NHC-ligated nickel(II) halides (Ni(NHC)2X2) (X = Cl, Br) with silver(I) and alkali metal cyanoborates were used to synthesize mono- and disubstituted coordination compounds of the type cis- or trans-[Ni(NHC)2(CB)X] (cis-10c, cis-11c, trans-12b) and cis- or trans-[Ni(NHC)2(CB)2] (cis-13b, cis-14a–c, trans-14a–b, trans-15b, trans-5b). Further investigations reveal that NHC-ligated cyanoborate complexes can act as building blocks for coordination polymers, as observed for structurally characterized ![]() trans‑[Ni(Mes2Im)2(µ2‑[NC‑BH2‑CN])2]·2Ag(µ2‑[BH2(CN)2])} (trans‑5b·Ag). This study demonstrates the diverse character of cyanoborates in coordination chemistry as both, non-coordinating counter-anions, and weakly to medium coordinating anions forming novel transition metal complexes and salts. It provides evidence that a proper choice of cyanoborate and a proper choice of co-ligand can lead to a rich coordination chemistry of cyanoborate anions.
trans‑[Ni(Mes2Im)2(µ2‑[NC‑BH2‑CN])2]·2Ag(µ2‑[BH2(CN)2])} (trans‑5b·Ag). This study demonstrates the diverse character of cyanoborates in coordination chemistry as both, non-coordinating counter-anions, and weakly to medium coordinating anions forming novel transition metal complexes and salts. It provides evidence that a proper choice of cyanoborate and a proper choice of co-ligand can lead to a rich coordination chemistry of cyanoborate anions.
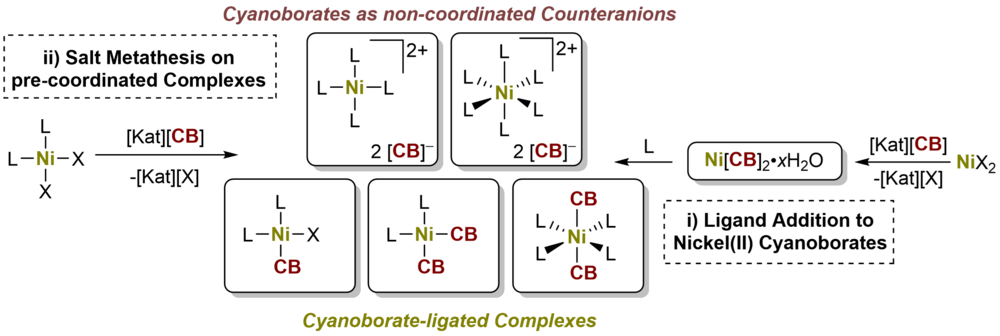
Nickel(II) Cyanoborates and Cyanoborate-Ligated Nickel(II) Complexes
M. S. Luff, C. Kerpen, J. A. P. Sprenger, M. Finze, U. Radius, Inorg. Chem. 2024, 63, 4, 2204-2216.
Nickel(II) cyanoborates Ni[BH2(CN)2]2·H2O (1b·H2O), Ni[BH(CN)3]2·0.5H2O (1c·0.5H2O), Ni[B(CN)4]2·0.5H2O (1d·0.5H2O) were synthesized and their reactivity with respect to dppeO2 (= 1,2-bis-(diphenylphosphinoethane dioxide)), pyNO (= pyridine-N-oxide), dppe (= 1,2-bis-(diphenylphosphinoethane)) and DMSO (= DMSO) was examined Using these ligands, either cyanoborate (CB) complex salts of [Ni(dppe)2]2+ (2b-d) and [Ni(pyNO)6]2+ (3c-d) were isolated or complexes [Ni(DMSO)4{NC-B(CN)3}2] (1dDMSO) and [Ni(dppeO2)2{NC-B(CN)3}2] (1ddppeO2) were formed. Salt metathesis of [Ni(dppe)Cl2] with alkali metal cyanoborates resulted in mono- and disubstituted coordination compounds [Ni(dppe){NC-BH(CN)2}Cl] (5c) and [Ni(dppe)(NC-BH2CN)2] (4b), which decomposed to the salts 2b-d. The synthetical pathways explored offer convenient routes to nickel(II) cyanoborates, nickel(II) complexes ligated with cyanoborates and nickel(II) complex salts of cyanoborates. Further, the authors' studies demonstrate the diverse character of cyanoborates in coordination chem. as noncoordinating counteranions on one side, but also as medium coordinating anions forming novel transition metal complexes and salts.
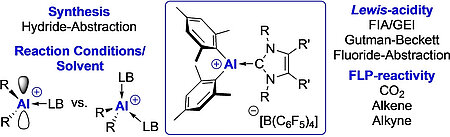
L. Werner, J. Hagn, J. Walpuski, U. Radius, Angew. Chem. Int. Ed. 2023, 62, e202312111.
The three-coordinate aluminum cations ligated by N-heterocyclic carbenes (NHCs) [(NHC) · AlMes2]+[B(C6F5)4]- (NHC=IMeMe4, IiPrMe5, IiPr 6, Mes=2,4,6-trimethylphenyl) were prepared via hydride abstraction of the alanes (NHC) · AlHMes2 (NHC=IMeMe1, IiPrMe2, IiPr 3) using [Ph3C]+[B(C6F5)4]- in toluene as hydride acceptor. If this reaction was performed in di-Et ether, the corresponding four-coordinate aluminum etherate cations [(NHC) · AlMes2(OEt2)]+ [B(C6F5)4]-7-9 (NHC=IMeMe7, IiPrMe8, IiPr 9) were isolated. According to a theor. and exptl. assessment of the Lewis-acidity of the [(IMeMe) · AlMes2]+ cation is the acidity larger than that of B(C6F5)3 and of similar magnitude as reported for Al(C6F5)3. The reaction of [(IMeMe) · AlMes2]+[B(C6F5)4]-4 with the sterically less demanding, basic phosphine PMe3 afforded a mixed NHC/phosphine stabilized cation [(IMeMe) · AlMes2(PMe3)]+[B(C6F5)4]-10. Equimolar mixtures of 4 and the sterically more demanding PCy3 gave a frustrated Lewis-pair (FLP), i.e., [(IMeMe) · AlMes2]+[B(C6F5)4]-/PCy3FLP-11, which reacts with small mols. such as CO2, ethene, and 2-butyne.
NHC-Stabilized Dialanes(4) of Al2Mes4
L. Werner, J. Hagn, U. Radius, Chem. Eur. J. 2023, 29, e202303111.
The synthesis and characterization of novel N-heterocyclic carbene (NHC) stabilized dialanes Al2Mes4 as well as first investigations concerning the reactivity of these compounds are reported. The synthesis of these compounds proceeds via the mesityl-substituted alanes (NHC)·AlHMes2 (NHC=IMeMe{=1,3,4,5-tetramethyl-imidazolin-2-ylidene}, IiPrMe {=1,3-di-iso-propyl-4,5-dimethylimidazolin-2-ylidene}) and iodo-alanes (NHC)·AlIMes2 (NHC=IMeMe, IiPrMe). Metallic reduction of (NHC)·AlIMes2 afforded the new NHC-stabilized dialanes (NHC)2·Al2Mes4 (NHC=IMeMe, IiPrMe). The NHC-ligated dialanes are thermally robust and storable synthons for the dialane Al2Mes4. First reactivity studies on (IMeMe)2·Al2Mes4 towards small mols. confirm this, as this compound shows controlled and selective reactions with several substrates. Reaction with CuCl leads to oxidation of the dialane and formation of (IMeMe)·AlClMes2, reactions with pyridine N-oxide and tBu-N=C=S, resp., gave the chalcogenide-bridged dimers {(IMeMe)·AlMes2}2-μ-E (E=O, S), and reaction with acetylene afforded the dimetallaacetylide {(IMeMe)·AlMes2}2-μ-(C≃C).
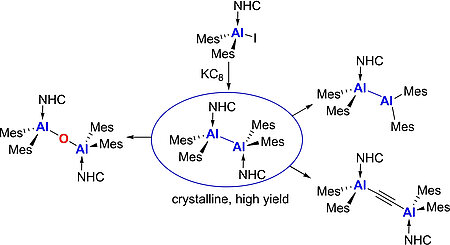
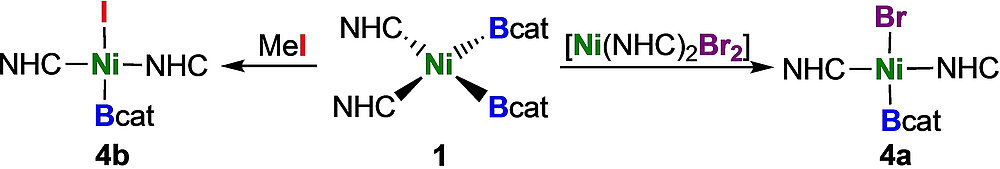
L. Tendera, L. Kuehn, T. B. Marder, U. Radius, Chem. Eur. J. 2023, 29, e202302310.
The synthesis of the first terminal mono-boryl complexes of nickel, which are not stabilized by a pincer ligand, is reported. The reaction of the nickel bis-boryl complex cis-[Ni(iPr2ImMe)2(Bcat)2] 1 (cat=1,2-O2C6H4) with the small donor ligand PMe3 led to a complete ligand exchange at nickel with reductive elimination of B2cat2 and formation of the bis-NHC adduct [B2cat2 · (iPr2ImMe)2] 3 and [Ni(PMe3)4] 2 as the metal-containing species. Electrophilic attack of MeI on complex 1 or ligand dismutation of 1 with trans-[Ni(iPr2ImMe)2Br2] led to loss of only one boryl ligand of 1 and afforded the nickel mono-boryl complexes trans-[Ni(iPr2ImMe)2(Bcat)Br] 4 a and trans-[Ni(iPr2ImMe)2(Bcat)I] 4 b.
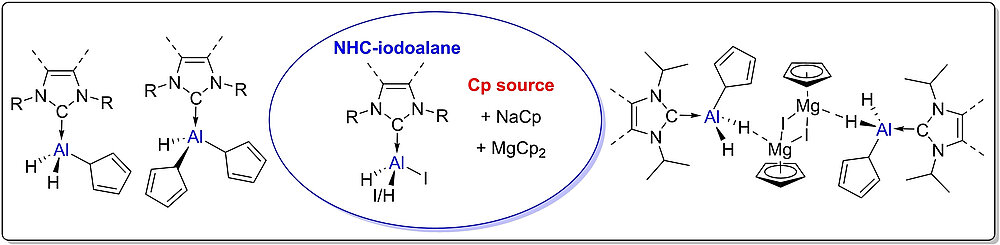
NHC-Adducts of Cyclopentadienyl-Substituted Alanes
L. Werner, S. Mann, U. Radius, Eur. J. Inorg. Chem. 2023, 26, e202300398.
The mono- and bis-iodo-substituted NHC-stabilized alanes (NHC) · AlH2I and (NHC) · AlHI2 offer a convenient entry for further substitution reactions at aluminum. Reactions of (NHC) · AlH2I 1-4 with one equivalent of NaCp afforded the adducts (NHC) · AlH2Cp 9-12 (NHC=Me2ImMe (9), iPr2ImMe (10), iPr2Im (11), Dipp2Im (12)). Alane adducts with two Cp substituents (NHC) · AlHCp213-16 (NHC=Me2ImMe (13), iPr2ImMe (14), iPr2Im (15), Dipp2Im (16)) were prepared by the analogous reaction of (NHC) · AlHI25-8 using two equivalent of NaCp. The unusual dimeric adducts ((NHC) · AlH2Cp · CpMgI)217-19 (NHC=Me2ImMe (17), iPr2ImMe (18), iPr2Im (19)) were obtained from the reaction of 1-3 with MgCp2.
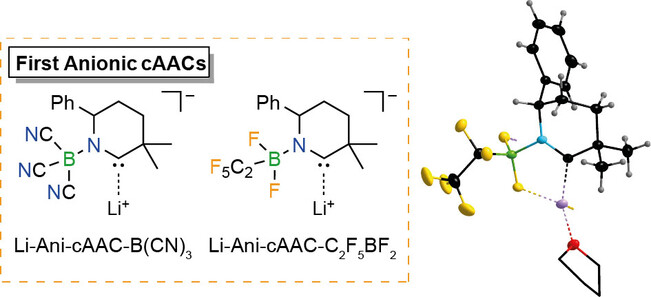
Boranes Paving the Way to Anionic Cyclic (Alkyl)(amino)carbenes (Ani-cAACs)
L. Zapf, S. Peters, U. Radius, M. Finze, Angew. Chem. Int. Ed. 2023, 62, e202300056.
First examples of anionic cyclic (alkyl)(amino)carbenes (Ani-cAACs) that contain borane substituents have been synthesized. The nature of the borane substituents allows a modulation of the σ-donor or π-acceptor abilities compared to their neutral analogs. A B(CN)3-substituted Ani-cAAC has been generated and used in situ. The corresponding C2F5BF2-Ani-cAAC 6 was obtained in high yield on a multigram scale. First reactions of these novel ligands with elemental selenium and chloro(triphenylphosphine)gold(I) led to the anionic selenium adducts 7 and 8 and the Ani-cAAC gold complex 9. The properties of these compounds and data derived from theor. calculations provide an insight into the electronic and steric properties of these novel anionic cAACs. Especially the ease of synthesis and the combination of properties such as neg. charge, large buried volume, and good σ-donor and π-acceptor ability renders Ani-cAACs unique and promising new building blocks.
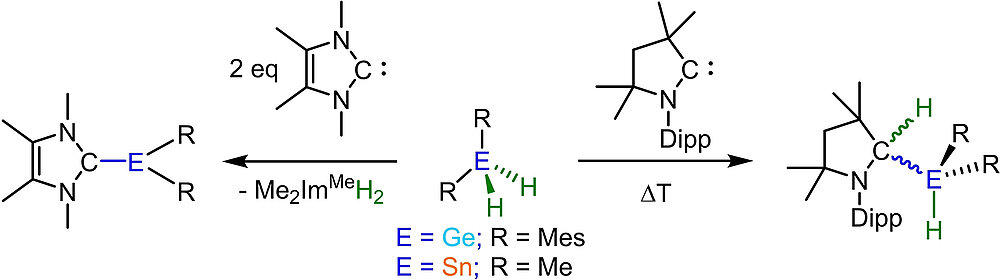
Activation of Ge-H and Sn-H Bonds with N-Heterocyclic Carbenes and a Cyclic (Alkyl)(amino)carbene
M. S. M. Philipp, R. Bertermann, U. Radius, Chem. Eur. J. 2023, 29, e202202493.
A study of the reactivity of several N-heterocyclic carbenes (NHCs) and the cyclic (alkyl)(amino)carbene 1-(2,6-di-iso-propylphenyl)-3,3,5,5-tetramethyl-pyrrolidin-2-ylidene (cAACMe) with the group 14 hydrides GeH2Mes2 and SnH2Me2 (Mes = 2,4,6-Me3C6H2) is presented. The reaction of GeH2Mes2 with cAACMe led to the insertion of cAACMe into one Ge-H bond to give cAACMeH-GeHMes2 (1). If 1,3,4,5-tetramethyl-imidazolin-2-ylidene (Me2ImMe) was used as the carbene, NHC-mediated dehydrogenative coupling occurred, which led to the NHC-stabilized germylene Me2ImMe·GeMes2 (2). The reaction of SnH2Me2 with cAACMe also afforded the insertion product cAACMeH-SnHMe2 (3), and reaction of two equiv Me2ImMe with SnH2Me2 gave the NHC-stabilized stannylene Me2ImMe·SnMe2 (4). If the sterically more demanding NHCs Me2ImMe, 1,3-di-isopropyl-4,5-dimethyl-imidazolin-2-ylidene (iPr2ImMe) and 1,3-bis-(2,6-di-isopropylphenyl)-imidazolin-2-ylidene (Dipp2Im) were employed, selective formation of cyclic oligomers (SnMe2)n (5; n = 5-8) in high yield was observed These cyclic oligomers were also obtained from the controlled decomposition of cAACMeH-SnHMe2 (3).
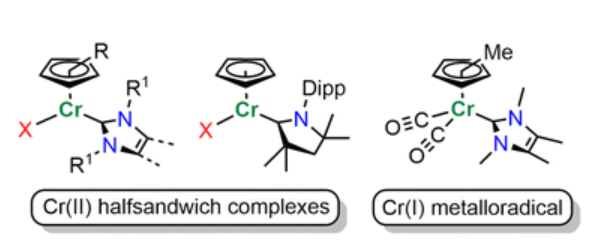
G. Horrer, M. S. Luff, U. Radius, Dalton Trans. 2023, 52, 13244-13257.
The synthesis and characterization of a series of Cr(II) N-Heterocyclic Carbene (NHC) complexes of the type [{Cr(NHC)Cl(μ-Cl)}2] and [(Cyp)Cr(NHC)X] (Cyp = η5-C5H5, cyclopentadienyl; η5-C5Me5, pentamethylcyclopentadienyl; X = Cl, η3-C3H5; NHC = IMeMe, IiPrMe, IMes, IDipp) as well as the cyclic (alkyl)(amino)carbene cAACMe ligated complexes [(η5-C5H5)Cr(cAACMe)X] (X = Cl, NPh2), [(η5-C9H7)Cr(cAACMe)Cl] (C9H7 = Ind, indenyl) and [(η5-C13H9)Cr(cAACMe)Cl] (C13H9 = Fl, fluorenyl) are reported. The reduction of [(η5-C5Me5)Cr(IMeMe)Cl] with KC8 in the presence of CO afforded the NHC ligated Cr(I) metallo-radical [(η5-C5Me5)Cr(IMeMe)(CO)2]. Quantum chem. calculations performed on [(η5-C5Me5)Cr(IMeMe)(CO)2] confirm for this complex a predominantly chromium centered radical.
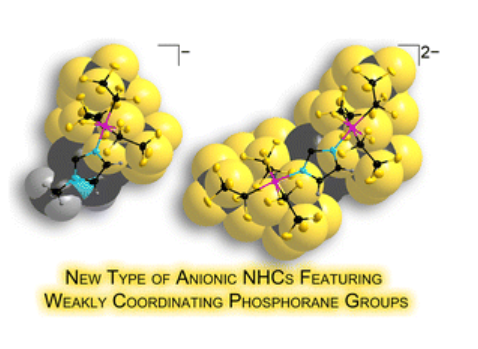
Anionic N-heterocyclic carbenes featuring weakly coordinating perfluoroalkylphosphorane moieties
L. Zapf, U. Radius, M. Finze, Dalton Trans., 2023, 52, 9553-9561.
The anionic 1-methyl-3-(tris(pentafluoroethyl)difluorophosphorane)imidazoline-2-ylidenate 3 and the 1,3-bis(tris(pentafluoroethyl)difluorophosphorane)imidazoline-2-ylidenate dianion 4 were obtained in high yield by deprotonation of {(C2F5)3PF2}-methylimidazole 1 and the {(C2F5)3PF2}2-imidazolate anion 2. Carbenes 3 and 4 are first examples for a novel class of NHCs carrying weakly coordinating anions (WCA-NHCs). First reactions of these new ligands with elemental selenium and chloro(phosphine)gold(I) complexes to result in an anionic selenium adduct (5) and WCA-NHC gold complexes (6 and 7) have been undertaken. The structural and spectroscopic properties of these NHC derivatives in conjunction with data from quantum chem. calculations provide an insight into the electronic and steric properties of the WCA-NHCs 3 and 4. Especially the combination of properties such as weakly coordinating periphery combined with the coordinative carbene center, neg. charge, large buried volume (%Vbur), and strong σ-donor as well as efficient π-acceptor ability render NHCs 3 and 4 unique and promising new ligands.
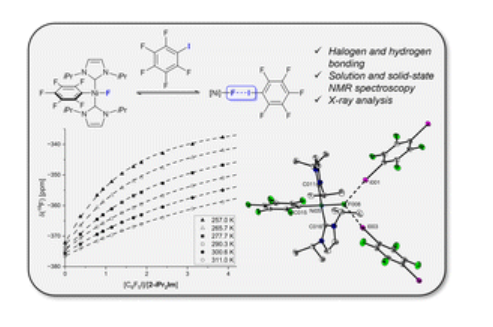
V. G. Thangavadivale, L. Tendera, R. Bertermann, U. Radius, T. Beweries, R. N. Perutz, Farad. Disc. 2023, 244, 62-76.
Nickel fluoride complexes of the type [Ni(F)(L)2(ArF)] (L = phosphine, ArF = fluorinated arene) are well-known to form strong halogen and hydrogen bonds in solution and in the solid state. A comprehensive study of such non-covalent interactions using bis(carbene) complexes as acceptors and suitable halogen and hydrogen bond donors is presented. In solution, the complex [Ni(F)(iPr2Im)2(C6F5)] forms halogen and hydrogen bonds with iodopentafluorobenzene and indole, respectively, which have formation constants (K300) an order of magnitude greater than those of structurally related phosphine supported nickel fluorides. Co-crystallisation of this complex and its backbone-methylated analogue [Ni(F)(iPr2Me2Im)2(C6F5)] with 1,4-diiodotetrafluorobenzene produces halogen bonding adducts which were characterised by X-ray analysis and 19F MAS solid state NMR analysis. Differences in the chemical shifts between the nickel fluoride and its halogen bonding adduct are well in line with data that were obtained from titration studies in solution.
Subvalent group 13 molecules by carbene-induced hydrogen abstraction
L. Werner, A. Hoch, C. Luz, M. Riethmann, U. Radius, Dalton Trans. 2023, 52, 7059-7070.
The N-Heterocyclic Carbene (NHC) alane and gallane adducts (NHC)·Cp*AlH2 (NHC = Me2ImMe5, iPr2ImMe6, Dipp2Im 7) and (NHC)·Cp*GaH2 (NHC = Me2ImMe8, iPr2ImMe9, Dipp2Im 10; R2Im = 1,3-di-organyl-imidazolin-2-ylidene; Dipp = 2,6 diisopropylphenyl; Me2ImMe = 1,3,4,5-tetra-methyl-imidazolin-2-ylidene; Cp* = C5Me5) were prepared either via the reaction of (AlH2Cp*)31 with the NHC or by the treatment of (NHC)·GaH2I (NHC = Me2ImMe2, iPr2ImMe3, Dipp2Im 4) with KCp*. The reaction of (AlH2Cp*)31 with the backbone saturated NHC Dipp2ImH led to NHC ring expansion instead with the formation of (RER-Dipp2ImHH2)AlCp* 12. Heating solutions of the gallium compounds 8–10 triggered reductive elimination of Cp*H and afforded Cp*GaI16. The reaction of the alane adduct (Me2ImMe)·Cp*AlH25 with cAACMe led to the insertion of cAACMe into the Al–H bond with the formation of the compound rac-(Me2ImMe)·AlHCp*(cAACMeH) rac-14. Heating a solution of rac-14 led to irreversible isomerisation with the formation of (Me2ImMe)·AlHCp*(cAACMeH) meso-14. The alane adducts (iPr2ImMe)·Cp*AlH26 and (Dipp2Im)·Cp*AlH27 react with cAACMe with the release of the NHC and formation of the exceptionally stable oxidative addition product (cAACMeH)AlHCp* 15. Reactions of the gallane adducts 8–10 with cAACMe led to reductive elimination of cAACMe–H2 and the formation of Cp*GaI16.
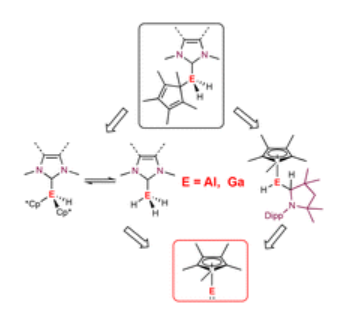
An easy-to-perform evaluation of steric properties of Lewis acids
L. Zapf, M. Riethmann, S. A. Föhrenbacher, M. Finze, U. Radius, Chem. Sci. 2023, 14, 2275-2288.
Steric and electronic effects play a very important role in chem., as these effects influence the shape and reactivity of mols. Herein, an easy-to-perform approach to assess and quantify steric properties of Lewis acids with differently substituted Lewis acidic centers is reported. This model applies the concept of the percent buried volume (%VBur) to fluoride adducts of Lewis acids, as many fluoride adducts are crystallog. characterized and are frequently calculated to judge fluoride ion affinities (FIAs). Thus, data such as cartesian coordinates are often easily available. A list of 240 Lewis acids together with topog. steric maps and cartesian coordinates of an oriented mol. suitable for the SambVca 2.1 web application is provided, together with different FIA values taken from the literature. Diagrams of %VBur as a scale for steric demand vs. FIA as a scale for Lewis acidity provide valuable information about stereo-electronic properties of Lewis acids and an excellent evaluation of steric and electronic features of the Lewis acid under consideration. Furthermore, a novel LAB-Rep model (Lewis acid/base repulsion model) is introduced, which judges steric repulsion in Lewis acid/base pairs and helps to predict if an arbitrary pair of Lewis acid and Lewis base can form an adduct with respect to their steric properties. The reliability of this model was evaluated in four selected case studies, which demonstrate the versatility of this model. For this purpose, a user-friendly Excel spreadsheet was developed and is provided in the ESI, which works with listed buried volumes of Lewis acids %VBur_LA and of Lewis bases %VBur_LB, and no results from exptl. crystal structures or quantum chem. calculations are necessary to evaluate steric repulsion in these Lewis acid/base pairs.
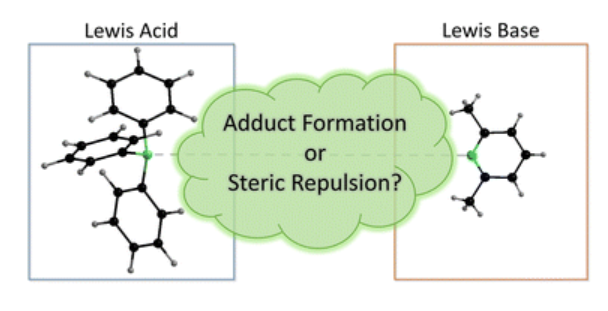
Nickel boryl complexes and nickel-catalyzed alkyne borylation
L. Tendera, F. Fantuzzi, T. B. Marder, U. Radius Chem. Sci. 2023, 14, 2215-2228.
The first nickel bis-boryl complexes cis-[Ni(iPr2ImMe)2(Bcat)2], cis-[Ni(iPr2ImMe)2(Bpin)2] and cis-[Ni(iPr2ImMe)2(Beg)2] are reported, which were prepared via the reaction of a source of [Ni(iPr2ImMe)2] with the diboron(4) compounds B2cat2, B2pin2 and B2eg2 (iPr2ImMe = 1,3-di-iso-propyl-4,5-dimethylimidazolin-2-ylidene; B2cat2 = bis(catecholato)diboron; B2pin2 = bis(pinacolato)diboron; B2eg2 = bis(ethylene glycolato)diboron). X-ray diffraction and DFT calculations strongly suggest that a delocalized, multicenter bonding scheme dictates the bonding situation of the NiB2 moiety in these square planar complexes, reminiscent of the bonding situation of “non-classical” H2 complexes. [Ni(iPr2ImMe)2] also efficiently catalyzes the diboration of alkynes using B2cat2 as the boron source under mild conditions. In contrast to the known platinum-catalyzed diboration, the nickel system follows a different mechanistic pathway, which not only provides the 1,2-borylation product in excellent yields, but also provides an efficient approach to other products such as C–C coupled borylation products or rare tetra-borylated compounds. The mechanism of the nickel-catalyzed alkyne borylation was examined by means of stoichiometric reactions and DFT calculations. Oxidative addition of the diboron reagent to nickel is not dominant; the first steps of the catalytic cycle are coordination of the alkyne to [Ni(iPr2ImMe)2] and subsequent borylation at the coordinated and, thus, activated alkyne to yield complexes of the type [Ni(NHC)2(η2-cis-(Bcat)(R)CC(R)(Bcat))], exemplified by the isolation and structural characterization of [Ni(iPr2ImMe)2(η2-cis-(Bcat)(Me)CC(Me)(Bcat))] and [Ni(iPr2ImMe)2(η2-cis-(Bcat)(H7C3)CC(C3H7)(BCat))].
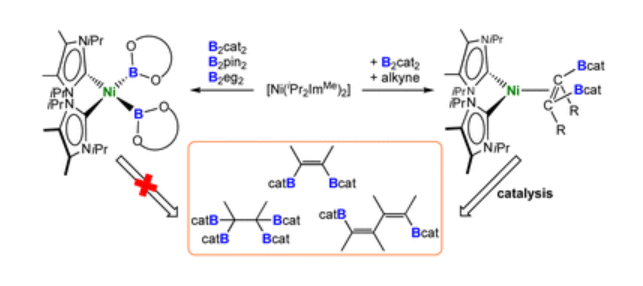
Michael S. M. Philipp, Dr. Mirjam J. Krahfuss, Dr. Krzysztof Radacki, Prof. Dr. Udo Radius Eur. J. Inorg. Chem. 2022, 28, e202202349.
A study on the reactivity of N-heterocyclic carbenes (NHCs) and the cyclic (alkyl)(amino)carbene cAACMe with selected germanium(IV) and tin(IV) chlorides and organyl chlorides is presented. The reactions of the NHCs Me2ImMe, iPr2ImMe and Dipp2Im with the methyl chlorides ECl2Me2 afforded the adducts NHC ⋅ ECl2Me2 (E=Ge (1), Sn (2)), NHC=Me2ImMe (a), iPr2ImMe (b), Dipp2Im (c)). The reaction of Me2ImMe with GeCl4 led to isolation of Me2ImMe ⋅ GeCl4 (3), the reaction of iPr2ImMe with SnCl4 in THF afforded the THF adduct iPr2ImMe ⋅ SnCl4 ⋅ THF (4). Dipp2Im ⋅ GeCl2Me2 (1 c) isomerized into the backbone coordinated imidazolium salt [aDipp2Im ⋅ GeClMe2][Cl] (5) upon thermal treatment. The reactions of cAACMe with (i) ECl2R2 (E=Ge, Sn) gave the adducts cAACMe ⋅ ECl2R2 (R=Me: E=Ge (6); Sn (7); Ph: E=Ge (8)), with (ii) GeClMe3 and GeCl4 the salts [cAACMe ⋅ GeMe3][Cl] (9) and [cAACMeCl][GeCl3] (10), and (iii) with SnCl4 the salt [cAACMeCl][SnCl3] (11) and the adduct cAACMe ⋅ SnCl4 (12). Reduction of 2 a with KC8 afforded the NHC-stabilized stannylene Me2ImMe ⋅ SnMe2 13, reduction of 7 with either KC8 or 1,4-bis-(trimethylsilyl)-1,4-dihydropyrazin in the presence of SnCl2Me2 yielded cAACMe ⋅ SnMe2 ⋅ SnMe2Cl2 (14).
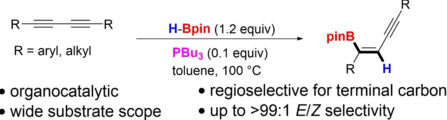
Swetha Jos, Connor Szwetkowski, Carla Slebodnick, Robert Ricker, Ka Lok Chan, Wing Chun Chan, Udo Radius, Zhenyang Lin, Todd B. Marder, Webster L. Santos, Chem. Eur. J. 2022, 28, e202202349.
We report a transition metal-free, regio- and stereoselective, phosphine-catalyzed method for the trans hydroboration of 1,3-diynes with pinacolborane that affords (E)-1-boryl-1,3-enynes. The reaction proceeds with excellent selectivity for boron addition to the external carbon of the 1,3-diyne framework as unambiguously established by NMR and X-ray crystallog. studies. The reaction displays a broad substrate scope including unsym. diynes to generate products in high yield (up to 95%). Exptl. and theor. studies suggest that phosphine attack on the alkyne is a key process in the catalytic cycle.
Cationic Nickel d9-Metalloradicals [Ni(NHC)2]+
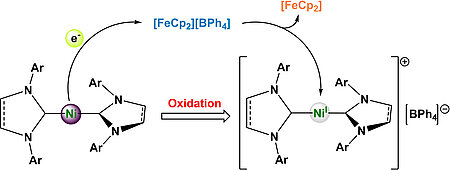
Lukas Tendera, Martin S. Luff, Ivo Krummenacher, Udo Radius, Eur. J. Inorg. Chem. 2022, e202200416.
A series of five new homoleptic, linear nickel d9-complexes of the type [NiI(NHC)2][BPh4] is reported. Starting from the literature known Ni(0) complexes [Ni(Mes2Im)2], [Ni(Mes2ImH2)2], [Ni(Dipp2Im)2], [Ni(Dipp2ImH2)2] and [Ni(cAACMe)2] (1-5; Mes2Im = 1,3-dimesityl-2-imidazolylidene, Mes2ImH2 = 1,3-dimesityl-2-imidazolidinylidene, Dipp2Im = 1,3-bis(2,6-diisopropylphenyl)-2-imidazolylidene, Dipp2ImH2 = 1,3-bis(2,6-diisopropylphenyl)-2-imidazolidinylidene, cAACMe = 1-(2,6-diisopropylphenyl)-3,3,5,5-tetramethyl-2-pyrrolidinylidene), their oxidized Ni(I) analogs [NiI(Mes2Im)2][BPh4] (1+), [NiI(Mes2ImH2)2][BPh4] (2+), [NiI(Dipp2Im)2][BPh4] (3+), [NiI(Dipp2ImH2)2][BPh4] (4+) and [NiI(cAACMe)2][BPh4] (5+) were synthesized by one-electron oxidation with ferrocenium tetraphenylborate. The complexes 1+-5+ were fully characterized including X-ray structure anal. The complex cations reveal linear geometries in the solid state and NMR spectra with extremely broad, paramagnetically shifted resonances. DFT calculations predicted an orbitally degenerate ground state leading to large magnetic anisotropy, which was verified by EPR measurements in solution and on solid samples. The magnetic anisotropy of the complexes is highly dependent from the steric protection of the metal atom, which results in a noticeable decrease of the g-tensor anisotropy for the N-Mes substituted complexes 1+ and 2+ in solution due to the formation of T-shaped THF adducts.
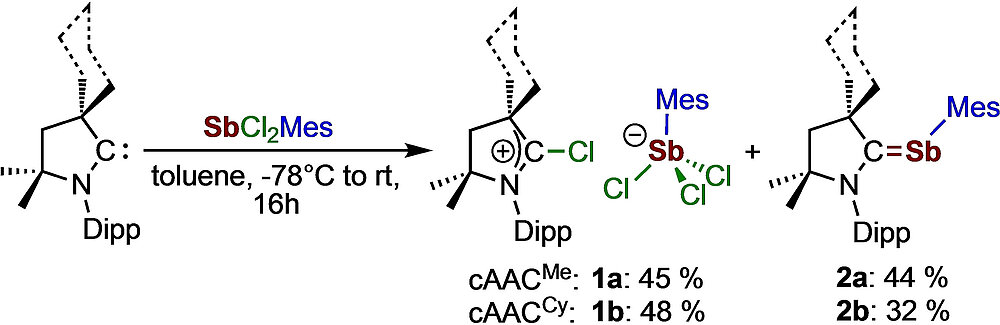
A Versatile Route To Cyclic (Alkyl)(Amino)Carbene-Stabilized Stibinidenes
Michael S. M. Philipp and Udo Radius, Z. Anorg. Allg. Chem. 2022, 648, e2022000.
A convenient route for the synthesis of the cAACMe (cAAC=cyclic (alkyl)(amino)carbene, cAACMe=1-(2,6-di-iso-propylphenyl)-3,3,5,5-tetramethyl-pyrrolidin-2-ylidene) and cAACCy (cAACCy=2-azaspiro[4.5]dec-2-(2,6-diisopropylphenyl)-3,3-dimethyl-1-ylidene) stabilized stibinidenes cAACMe⋅SbMes (2 a) (Mes=2,4,6-trimethylphenyl) and cAACCy⋅SbMes (2 b) is reported. A mechanism for the formation of [cAACRCl][SbCl3Mes] 1 and cAACR⋅SbMes 2 from the reaction of cAAC with the antimony(III) precursor SbCl2Mes, which proceeds via the isolable intermediate [cAACRSbClMes][SbCl3Mes] (3), is proposed.
Ludwig Zapf, Sven Peters, Rüdiger Bertermann, Udo Radius* and Maik Finze*, Chem. Eur. J. 2022, 28, e202200275.
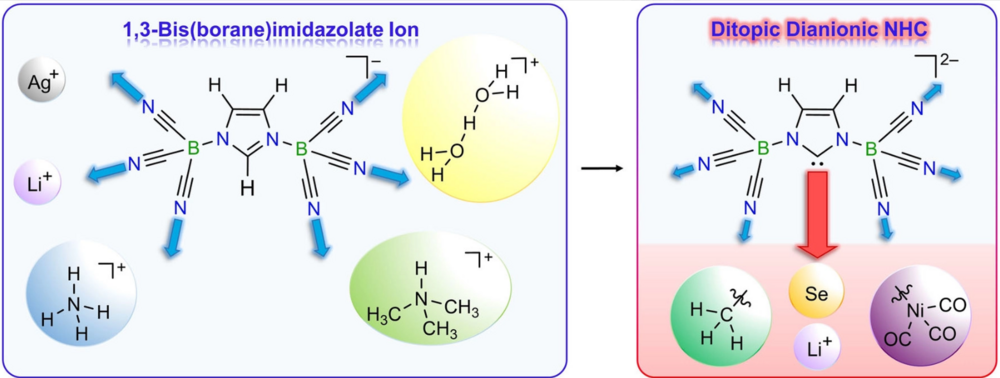
The 1-methyl-3-(tricyanoborane)imidazolin-2-ylidenate anion (2) was obtained in high yield by deprotonation of the B(CN)3-methylimidazole adduct 1. Regarding charge and stereo-electronic properties, anion 2 closes the gap between well-known neutral NHCs and the ditopic dianionic NHC, the 1,3-bis(tricyanoborane)imidazolin-2-ylidenate dianion (IIb). The influence of the number of N-bonded tricyanoborane moieties on the σ-donating and π-accepting properties of NHCs was assessed by quantum chemical calculations and verified by experimental data on 2, IIb, and 1,3-dimethylimidazolin-2-ylidene (IMe, IIa). Therefore NHC 2, which acts as a ditopic ligand via the carbene center and the cyano groups, was reacted with alkyl iodides, selenium, and [Ni(CO)4] yielding alkylated imidazoles 3 and 4, the anionic selenium adduct 5, and the anionic nickel tricarbonyl complex 8, respectively. The results of this study prove that charge, number of coordination sites, buried volume (%Vbur) and σ-donor and π-acceptor abilities of NHCs can be effectively fine-tuned via the number of tricyanoborane substituents.
Selective, Transition Metal-free 1,2-Diboration of Alkyl Halides, Tosylates, and Alcohols
Mingming Huang, Dr. Jiefeng Hu, Shasha Shi, Dr. Alexandra Friedrich, Johannes Krebs, Prof. Dr. Stephen A. Westcott, Prof. Dr. Udo Radius, Prof. Dr. Todd B. Marder, Chem. Eur. J. 2022, 28, e202200480.
Defunctionalization of readily available feedstocks to provide alkenes for the synthesis of multifunctional mols. represents an extremely useful process in organic synthesis. Herein, authors describe a transition metal-free, simple and efficient strategy to access alkyl 1,2-bis(boronate esters) via regio- and diastereoselective diboration of secondary and tertiary alkyl halides (Br, Cl, I), tosylates, and alcs. Control experiments demonstrated that the key to this high reactivity and selectivity is the addition of a combination of potassium iodide and N,N-dimethylacetamide (DMA). The practicality and industrial potential of this transformation are demonstrated by its operational simplicity, wide functional group tolerance, and the late-stage modification of complex mols. From a drug discovery perspective, this synthetic method offers control of the position of diversification and diastereoselectivity in complex ring scaffolds, which would be especially useful in a lead optimization program.

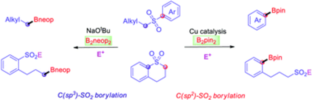
Base-Mediated Radical Borylation of Alkyl Sulfones
Mingming Huang, Dr. Jiefeng Hu, Dr. Ivo Krummenacher, Dr. Alexandra Friedrich, Prof. Dr. Holger Braunschweig, Prof. Dr. Stephen A. Westcott, Prof. Dr. Udo Radius, Prof. Dr. Todd B. Marder, Chem. Eur. J. 2022, 28, e202103866.
A practical and direct method was developed for the production of versatile alkyl boronate esters via transition metal-free borylation of primary and secondary alkyl sulfones. The key to the success of the strategy is the use of bis(neopentyl glycolato) diboron (B2neop2), with a stoichiometric amount of base as a promoter. The practicality and industrial potential of this protocol are highlighted by its wide functional group tolerance, the late-stage modification of complex compounds, no need for further transesterification, and operational simplicity. Radical clock, radical trap experiments, and EPR studies were conducted which show that the borylation process involves radical intermediates.
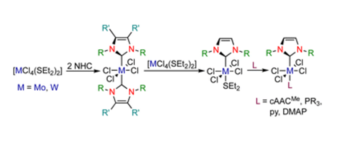
N-heterocyclic carbene and cyclic (alkyl)(amino)carbene complexes of molybdenum(iv) and tungsten(iv)
Christian Luz, Eduard Glok, Günther Horrer and Udo Radius*, Dalton Trans., 2022, 51, 18337.
The synthesis and characterization of N-heterocyclic carbene (NHC) and cyclic (alkyl)(amino)carbene (cAAC) complexes of molybdenum(IV) and tungsten(IV) chloride is reported. Reaction of two equivalents of the NHCs IMes (1,3-bis(2,4,6-trimethylphenyl)imidazolin-2-ylidene), IDipp (1,3-bis(2,6-diisopropylphenyl)imidazolin-2-ylidene) and IiPrMe (1,3-diisopropyl-4,5-dimethylimidazolin-2-ylidene) with [MCl4(SEt)2] (M = Mo, W) in toluene afforded the bis-NHC complexes [MCl4(NHC)2] (M = Mo: NHC = IMes 1, NHC = IMes 2; M = W: NHC = IMes 3, NHC = IDipp 4, NHC = IiPrMe5). Mono-carbene complexes [MCl4(NHC)(SEt2)] 6–9 (M = Mo: NHC = IMes 6, NHC = IMes 7; M = W: NHC = IMes 8, NHC = IDipp 9) are available via ligand dismutation of 1–4 with [MCl4(SEt)2]. Complexes 1–9 were characterized by using elemental analysis, IR- and multinuclear NMR spectroscopy and X-ray diffraction for 1–8. The reactivity of the mono-NHC complexes 6–9 towards the cyclic (alkyl)(amino)carbene cAACMe as well as the two electron donor ligands trimethylphosphine, triphenylphosphine, pyridine (py) and N,N-dimethylpyridin-4-amine (DMAP) was studied, which led to the synthesis of the mixed substituted complexes [MCl4(NHC)(cAACMe)] (M = Mo: NHC = IMes 10, NHC = IDipp 11; M = W: NHC = IMes 12, NHC = IDipp 13), [MCl4(NHC)(PR3)] (M = W: NHC = IMes, R = Me 14, NHC = IMes, R = Ph 15, NHC = IDipp, R = Me 16, NHC = IDipp, R = Ph 17), [MCl4(NHC)(py)] (M = Mo: NHC = IMes 18, NHC = IDipp 20; M = W: NHC = IMes 22, NHC = IDipp 24) and [MCl4(NHC)(DMAP)] (M = Mo: NHC = IMes 19, NHC = IDipp 21; M = W: NHC = IMes 23, NHC = IDipp 25).
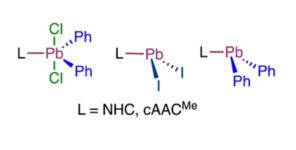
N-heterocyclic carbene and cyclic (alkyl)(amino)carbene adducts of plumbanes and plumbylenes
Michael S. M. Philipp, Rüdiger Bertermann and Udo Radius*, Dalton Trans., 2022, 51, 13488.
Lewis-acid/base adducts of N-heterocyclic carbenes (NHCs) and the cyclic (alkyl)(amino)carbene cAACMe (1-(2,6-di-iso-propylphenyl)-3,3,5,5-tetramethyl-pyrrolidin-2-ylidene) with selected lead(II) and lead(IV) compounds are presented. The reaction of the NHCs Me2ImMe (1,3,4,5-tetramethyl-imidazolin-2-ylidene), iPr2ImMe (1,3-di-isopropyl-4,5-dimethyl-imidazolin-2-ylidene), Dipp2Im (1,3-bis-(2,6-di-isopropylphenyl)-imidazolin-2-ylidene) and cAACMe (1-(2,6-di-iso-propylphenyl)-3,3,5,5-tetramethyl-pyrrolidin-2-ylidene) with PbI2 yielded the NHC-containing plumbylenes NHC·PbI2 (NHC = Me2ImMe (1), iPr2ImMe (2), Dipp2Im (3) and cAACMe·PbI2 (4)). Using the Pb(IV) compound PbCl2Ph2, the plumbane adducts NHC·PbCl2Ph2 (NHC = Me2ImMe (5), iPr2ImMe (6), Dipp2Im (7)) and cAACMe·PbCl2Ph2 (8)) were isolated in high yields. Reduction of the lead(IV) adducts 5 and 6 with excess KC8 afforded the diaryl substituted plumbylenes Me2ImMe·PbPh2 (9) and iPr2ImMe·PbPh2 (10), which are stable in the solid state but decompose in solution.
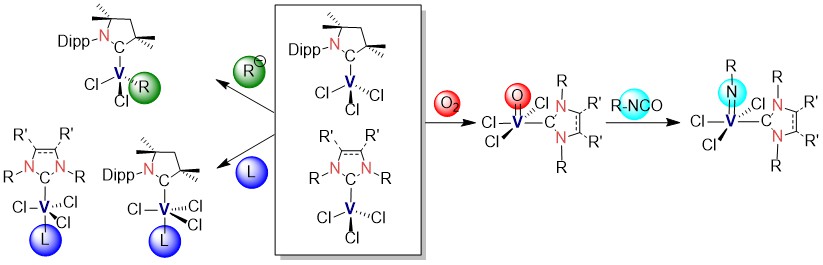
N-Heterocyclic carbene and cyclic (alkyl)(amino)carbene complexes of vanadium(III) and vanadium(V)
Günther Horrer, Ivo Krummenacher, Sophie Mann, Holger Braunschweig, and Udo Radius, Dalton Trans. 2022, 51, 11054.
[VCl3(THF)3] offers a convenient entrance point into the chem. of carbene stabilized V(III) complexes. Herein we report the paramagnetic mono- and biscarbene complexes [VCl3(cAAC)] (1, cAAC = 1-Dipp-3,3,5,5-tetramethyl-2-pyrrolidinylidene), [VCl3(cAAC)(THF)] (1-thf), [VCl3(IMes)] (2), [{VCl2(IiPrMe)(μ-Cl)}2] (3, IiPrMe = 1,3-diisopropyl-4,5-dimethyl-2-imidazolylidene), [VCl3(IDipp)] (4) [VCl3(SIDipp)] (5), [VCl3(SIDipp)(THF)] (5-thf), [VCl3(ItBu)] (6, ItBu = 1,3-di-tert-butyl-2-imidazolylidene), [VCl3(cAAC)2] (7) and [VCl3(IiPrMe)2] (8). Reaction of 1 with MesMgCl, MesLi and LiNPh2 afforded the complexes [VCl2(Mes)(cAACMe)] (9), [cAAC-H]+[VCl2Mes2]- (10) and [VCl2(NPh2)(cAAC)] (11). The vanadium(V) complexes [V(O)Cl3(IDipp)] (12) and [V(O)Cl3(SIDipp)] (13) were selectively prepared from oxygen oxidation of 4 and 5. The complex 12 and [V(O)Cl3(IMes)] react with isocyanates ArNCO to yield the NHC-ligated imido complexes [V(:NAr1)Cl3(IDipp)] (14, Ar1 = 4-MeC6H4), [V(:NAr2)Cl3(IDipp)] (15, Ar2 = 4-FC6H4), [V(:NAr1)Cl3(SIDipp)] (16), [V(:NAr2)Cl3(SIDipp)] (17), [V(:NAr1)Cl3(IMes)] (18) and [V(:NAr2)Cl3(IMes)] (19).
NHC induced radical formation via homolytic cleavage of B–B bonds and its role in organic reactions
Laura Kuehn, Ludwig Zapf, Luis Werner, Martin Stang, Sabrina Wurtemberger-Pietsch, Ivo Krummenacher, Holger Braunschweig, Emmanuel Lacote, Todd B. Marder * and Udo Radius *, Chem. Sci., 2022, 13, 8321.
New borylation methodologies have been reported recently, wherein diboron(4) compounds apparently participate in free radical couplings via the homolytic cleavage of the B–B bond. We report herein that bis-NHC adducts of the type (NHC)2$B2(OR)4, which are thermally unstable and undergo intramolecular ring expansion reactions (RER), are sources of boryl radicals of the type NHC–BR2c, exemplified by Me2ImMe$Bneopc 1a (Me2ImMe ¼ 1,3,4,5-tetramethyl-imidazolin-2-ylidene, neop ¼ neopentylglycolato), which are formed by homolytic B–B bond cleavage. Attempts to apply the boryl moiety 1a in a metalfree borylation reaction by suppressing the RER failed. However, based on these findings, a protocol was developed using Me2ImMeB2pin2 3 for the transition metal- and additive-free boryl transfer to substituted aryl iodides and bromides giving aryl boronate esters in good yields. Analysis of the side products and further studies concerning the reaction mechanism revealed that radicals are likely involved. An aryl radical was trapped by TEMPO, an EPR resonance, which was suggestive of a boronbased radical, was detected in situ, and running the reaction in styrene led to the formation of polystyrene. The isolation of a boronium cation side product, [(Me2ImMe)2Bpin]+I 7, demonstrated the fate of the second boryl moiety of B2pin2. Interestingly, Me2ImMe NHC reacts with aryl iodides and bromides generating radicals. A mechanism for the boryl radical transfer from Me2ImMeB2pin2 3 to aryl iodides and bromides is proposed based on these experimental observations.
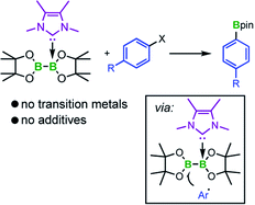
Cu-mediated vs. Cu-free selective borylation of aryl alkyl sulfones
Huang, Mingming; Tang, Man; Hu, Jiefeng; Westcott, Stephen A.; Radius, Udo; Marder, Todd B., Chem. Commun. 2022, 58(3), 395-398.
A Cu-catalyzed borylation of aryl alkyl sulfones was developed for the high yield synthesis of versatile arylboronic esters using a readily prepared NHC-Cu catalyst. In addition, the selective cleavage of either alkyl(C)-sulfonyl or aryl(C)-sulfonyl bonds of a cyclic sulfone via Cu-free or Cu-mediated processes generates the corresponding sulfinate salts, which can be further derivatized to provide sulfonyl-containing boronate esters, such as sulfones and sulfonyl fluorides.
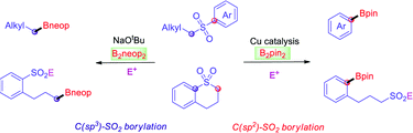
Zhiqiang Liu, Goutam Kumar Kole, Yudha P. Budiman, Ya-Ming Tian, Alexandra Friedrich, Xiaoling Luo,* Stephen A. Westcott, Udo Radius,* and Todd B. Marder*, Angew. Chem. Int. Ed. 2021, 60, 16529–16538.
A novel protocol for the transition metal-free 1,2-addition of polyfluoroaryl boronate esters to aldehydes and ketones is reported, which provides secondary alcohols, tertiary alcohols, and ketones. Control experiments and DFT calculations indicate that both the ortho-F substituents on the polyfluorophenyl boronates and the counterion K+ in the carbonate base are critical. The distinguishing features of this procedure include the employment of commercially available starting materials and the broad scope of the reaction with a wide variety of carbonyl compounds giving moderate to excellent yields. Intriguing structural features involving O−H⋅⋅⋅O and O−H⋅⋅⋅N hydrogen bonding, as well as arene-perfluoroarene interactions, in this series of racemic polyfluoroaryl carbinols have also been addressed.
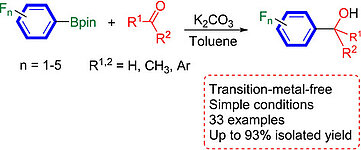
Zhi-Feng Jiao, Ya-Ming Tian, Xiao-Ning Guo, Udo Radius , Holger Braunschweig , Todd B. Marder , Xiang-Yun Guo, J. Cat. 2021, 395, 258–265.
A highly efficient photocatalytic protocol for borylation of alkyl bromides and chlorides with graphene supported Cu/Pd alloy nanoparticles as a heterogeneous catalyst is reported. This photocatalytic system operates with visible light in air, providing a wide range of primary and secondary alkyl halides with B2pin2 or B2neop2 in high yields at low temperatures, thereby demonstrating its broad utility and functional group tolerance. The high performance is attributed to a synergistic effect of localized surface plasmon resonance (LSPR) of Cu and charge transfer from Cu to Pd due to the alloy surface charge heterogeneity. Transfer of energetic electrons from Pd to electrophilic alkyl halides lead to the formation of the alkyl radicals, which quickly react with a nucleophilic adduct of a diboron compound with base adsorbed on the positively charged Cu sites to form the corresponding borylation product.
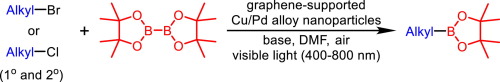
Photoinduced Borylation for the Synthesis of Organoboron Compounds
Ya-Ming Tian, Xiao-Ning Guo,* Holger Braunschweig, Udo Radius,* and Todd B. Marder*, Chem. Rev. 2021, 121, 3561−359.
Organoboron compounds have important synthetic value and can be applied in numerous transformations. The development of practical and convenient ways to synthesize boronate esters has thus attracted significant interest. Photoinduced borylations originated from stoichiometric reactions of alkanes and arenes with well-defined metal–boryl complexes. Now, photoredox-initiated borylations, catalyzed by either transition metal or organic photocatalysts, and photochemical borylations with high efficiency have become a burgeoning area of research. In this Focus Review, we summarize research on photoinduced borylations, especially emphasizing recent developments and trends. This includes the photoinduced borylation of arenes, alkanes, aryl/alkyl halides, activated carboxylic acids, amines, alcohols, and so on based on transition metal catalysis, metal-free organocatalysis, and direct photochemical activation. We focus on reaction mechanisms involving single-electron transfer, triplet-energy transfer, and other radical processes.
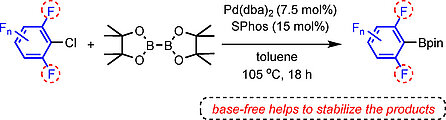
Base-Free Pd-Catalyzed C-Cl Borylation of Fluorinated Aryl Chlorides
Yudha P. Budiman, Sabine Lorenzen, Zhiqiang Liu, Udo Radius,* and Todd B. Marder*, Chem. Eur. J. 2021, 27, 3869 –3874.
Catalytic C−X borylation of aryl halides containing two ortho-fluorines has been found to be challenging, as most previous methods require stoichiometric amounts of base and the polyfluorinated aryl boronates suffer from protodeboronation, which is accelerated by ortho-fluorine substituents. Herein, we report that a combination of Pd(dba)2 (dba=dibenzylideneacetone) with SPhos (2-dicyclohexylphosphino-2’,6’-dimethoxybiphenyl) as a ligand is efficient to catalyze the C-Cl borylation of aryl chlorides containing two ortho-fluorine substituents. This method, conducted under base-free conditions, is compatible with the resulting di-ortho-fluorinated aryl boronate products which are sensitive to base.
Fluorinated Aryl Boronates as Building Blocks in Organic Synthesis
Yudha P. Budiman, Stephen A. Westcott, Udo Radius* and Todd B. Marder, Adv. Synth. Catal. 2021, 363, 2224 – 2255.
Organoboron compounds are well known building blocks for many organic reactions. However, under basic conditions, polyfluorinated aryl boronic acid derivatives suffer from instability issues that are accelerated in compounds containing an ortho-fluorine group, which result in the formation of the corresponding protodeboronation products. Therefore, a considerable amount of research has focused on novel methodologies to synthesize these valuable compounds while avoiding the protodeboronation issue. This review summarizes the latest developments in the synthesis of fluorinated aryl boronic acid derivatives and their applications in cross-coupling reactions and other transformations.
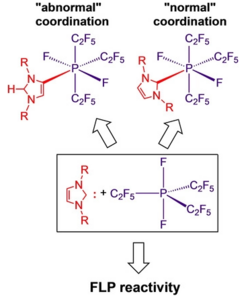
Steffen A. Föhrenbacher, Vivien Zeh, Mirjam J. Krahfuss, Nikolai V. Ignat’ev, Maik Finze,* and Udo Radius*, Eur. J. Inorg. Chem. 2021, 1941–1960.
The synthesis and characterization of Lewis acid/base adducts between tris(pentafluoroethyl)difluorophosphorane PF2(C2F5)3 and selected N-heterocyclic carbenes (NHCs) R2Im (1,3-di-organyl-imidazolin-2-ylidene) and phosphines are reported. For NHCs with small alkyl substituents at nitrogen (R=Me, nPr, iPr) the adducts NHC ⋅ PF2(C2F5)3 (2 a–h) were isolated. The reaction with the sterically more demanding NHCs Dipp2Im (1,3-bis-(2,6-di-iso-propylphenyl)-imidazolin-2-ylidene) (1 i) and tBu2Im (1,3-di-tert-butyl-imidazolin-2-ylidene) (1 j) afforded the aNHC adducts 3 i and 3 j (a denotes “abnormal” NHC coordination via a backbone carbon atom). The use of tBuMeIm (1-tert-butyl-3-methyl-imidazolin-2-ylidene) (1 m) led to partial decomposition of the NHC and formation of the salt [tBuMeIm−H][MeIm ⋅ PF2(C2F5)3] (4 m). The phosphorane PF2(C2F5)3 forms adducts with PMe3 but does not react with PPh3 or PCy3. The mer-cis isomer of literature-known Me3P ⋅ PF2(C2F5)3 (5 a) was structurally characterized. Mixtures of the phosphorane PF2(C2F5)3 and the sterically encumbered NHCs tBu2Im, Dipp2Im, and Dipp2ImH2 (1,3-bis-(2,6-di-iso-propylphenyl)-imidazolidin-2-ylidene) (1 k) showed properties of FLPs (Frustrated Lewis Pairs) as these mixtures were able to open the ring of THF (tetrahydrofuran) to yield NHC−(CH2)4O−PF2(C2F5)3 6 i–k. Furthermore, the deprotonation of the weak C−H acids CH3CN, acetone, and ethyl acetate was achieved, which led to the formation of the corresponding imidazolium salts and the phosphates [PF2(C2F5)3(CH2CN)]− (7), [PF2(C2F5)3(OC(=CH2)CH3)]− (8) and [PF2(C2F5)3(CH2CO2Et)]− (9).
Steffen A. Föhrenbacher, Mirjam J. Krahfuss, Ludwig Zapf, Alexandra Friedrich, Nikolai V. Ignat’ev, Maik Finze,* and Udo Radius*, Chem. Eur. J. 2021, 27, 3504 –3516.
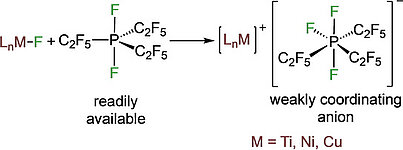
Fluoride abstraction from different types of transition metal fluoride complexes [LnMF] (M=Ti, Ni, Cu) by the Lewis acid tris(pentafluoroethyl)difluorophosphorane (C2F5)3PF2 to yield cationic transition metal complexes with the tris(pentafluoroethyl)trifluorophosphate counterion (FAP anion, [(C2F5)3PF3]−) is reported. (C2F5)3PF2 reacted with trans-[Ni(iPr2Im)2(ArF)F] (iPr2Im=1,3-diisopropylimidazolin-2-ylidene; ArF=C6F5, 1 a; 4-CF3-C6F4, 1 b; 4-C6F5-C6F4, 1 c) through fluoride transfer to form the complex salts trans-[Ni(iPr2Im)2(solv)(ArF)]FAP (2 a-c[solv]; solv=Et2O, CH2Cl2, THF) depending on the reaction medium. In the presence of stronger Lewis bases such as carbenes or PPh3, solvent coordination was suppressed and the complexes trans-[Ni(iPr2Im)2(PPh3)(C6F5)]FAP (trans-2 a[PPh3]) and cis-[Ni(iPr2Im)2(Dipp2Im)(C6F5)]FAP (cis-2 a[Dipp2Im]) (Dipp2Im=1,3-bis(2,6-diisopropylphenyl)imidazolin-2-ylidene) were isolated. Fluoride abstraction from [(Dipp2Im)CuF] (3) in CH2Cl2 or 1,2-difluorobenzene led to the isolation of [{(Dipp2Im)Cu}2]2+2 FAP− (4). Subsequent reaction of 4 with PPh3 and different carbenes resulted in the complexes [(Dipp2Im)Cu(LB)]FAP (5 a–e, LB=Lewis base). In the presence of C6Me6, fluoride transfer afforded [(Dipp2Im)Cu(C6Me6)]FAP (5 f), which serves as a source of [(Dipp2Im)Cu)]+. Fluoride abstraction of [Cp2TiF2] (7) resulted in the formation of dinuclear [FCp2Ti(μ-F)TiCp2F]FAP (8) (Cp=η5-C5H5) with one terminal fluoride ligand at each titanium atom and an additional bridging fluoride ligand.
NON-Ligated N-Heterocyclic Tetrylenes
Felix Krämer, Martin S. Luff, Udo Radius, Florian Weigend, and Frank Breher, Eur. J. Inorg. Chem. 2021, 3591–3600.
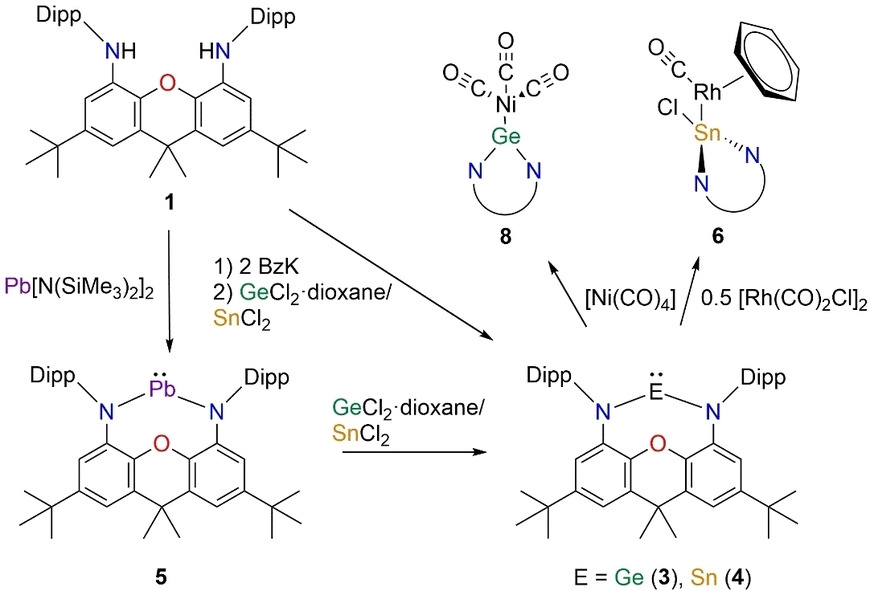
We report on the synthesis of N-heterocyclic tetrylenes ligated by the NON-donor framework 4,5-bis(2,6-diisopropylphenyl-amino)-2,7-di-tert-butyl-9,9-dimethylxanthene. The molecular structures of the germylene (3), stannylene (4) and plumbylene (5) where determined by X-ray diffraction studies. Furthermore, we present quantum chemical studies on the σ-donor and π-acceptor properties of 3–5. Additionally, we report on the reactivity of the tetrylenes towards the transition metal carbonyls [Rh(CO)2Cl]2, [W(CO)6] and [Ni(CO)4]. The isolated complexes (6 and 7) show the differing reactivity of NHTs compared to NHCs. Instead of just forming the anticipated complex [(NON)Sn−Rh(CO)2Cl], 4 inserts into the Rh−Cl bond to afford [(NON)Sn(Cl)Rh(CO)(C6H6)] (6, additional CO/C6H6 exchange) and [(NON)Sn(Cl)Rh2(CO)4Cl] (7). By avoiding halogenated transition metal precursors in order to prevent insertion reactions, germylene 3 shows “classical” coordination chemistry towards {Ni(CO)3} forming the complex [(NON)Ge−Ni(CO)3] (8).
N-Heterocyclic Carbene and Cyclic (Alkyl)(amino)carbene Adducts of Antimony(III)
Michael S. M. Philipp, Mirjam J. Krahfuss, Krzysztof Radacki, and Udo Radius, Eur. J. Inorg. Chem. 2021, 4007–4019.
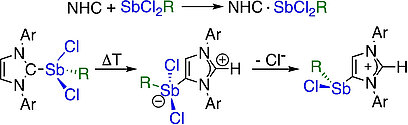
A systematic study on Lewis-acid/base adducts of N-heterocyclic carbenes (NHCs) and the cyclic (alkyl)(amino)carbene cAACMe (1-(2,6-di-iso-propylphenyl)-3,3,5,5-tetramethyl-pyrrolidin-2-ylidene) with antimony(III) chlorides of the general formula SbCl2R (R=Cl, Ph, Mes) is presented. The reaction of the NHCs Me2ImMe (1,3,4,5-tetra-methyl-imidazolin-2-ylidene), iPr2ImMe (1,3-di-isopropyl-4,5-dimethyl-imidazolin-2-ylidene), Mes2Im, Dipp2Im (R2Im=1,3-di-organyl-imidazolin-2-ylidene; Mes=2,4,6-trimethylphenyl, Dipp=2,6-di-isopropylphenyl) and cAACMe with antimony(III) compounds SbCl2R (R=Cl (1), Ph (2) and Mes (3)) yields the adducts NHC ⋅ SbCl2R (R=Cl (4), Ph (5) and Mes (6); NHC=Me2ImMe (a), iPr2ImMe (b), Dipp2Im (c) and Mes2Im (d)) and cAACMe ⋅ SbCl2R (R=Cl (4 e) and Ph (5 e)). Thermal treatment of (Dipp2Im) ⋅ SbCl2Ar (Ar=Ph (5 c) and Mes (6 c)) in benzene leads to isomerization to the backbone coordinated aNHC-adduct aDipp2Im ⋅ SbCl2Ar (Ar=Mes (7) and Ph (8)) (“a” denotes “abnormal” coordination mode of the NHC) in high yields. One of the chloride substituents at antimony of 7 can be abstracted by GaCl3 or Ag[BF4] to obtain the imidazolium salts [aDipp2Im ⋅ SbClMes][BF4] (9) and [aDipp2Im ⋅ SbClMes][GaCl4] (10).
Mirjam J. Krahfuss and Udo Radius, Eur. J. Inorg. Chem. 2021, 548–561.
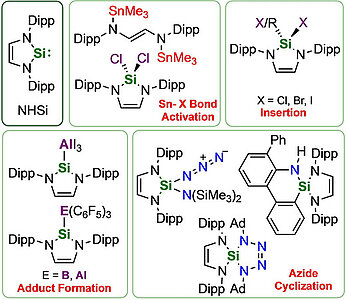
Investigations concerning the reactivity of the N-heterocyclic silylene Dipp2NHSi (1, 1,3-bis(2,6-diisopropylphenyl)-1,3-diaza-2-silacyclopent-4-en-2-ylidene) towards selected alanes and boranes, elemental halides X2 (X=Br, I), selected halide containing substrates such as tin chlorides and halocarbons, as well as organoazides are presented. The NHSi adducts Dipp2NHSi⋅AlI3 (2), Dipp2NHSi⋅Al(C6F5)3 (3), and Dipp2NHSi⋅B(C6F5)3 (4) were formed by the reaction of Dipp2NHSi with the corresponding Lewis acids AlI3, Al(C6F5)3 and B(C6F5)3. Adducts 3 and 4 were tested with respect to their ability to activate small organic molecules, but no frustrated Lewis pair reactivity was observed. Reactions of Dipp2NHSi with Br2, I2, Ph2SnCl2 and Me3SnCl led to formation of Dipp2NHSiBr2 (5), Dipp2NHSiI2 (6), Dipp2NHSiCl2 (7) and {(Me3Sn)N(Dipp)CH}2 (8), respectively. The reaction with the halocarbons methyl iodide, benzyl chloride, and benzyl bromide afforded the insertion products Dipp2NHSi(I)(CH3) (9), Dipp2NHSi(Cl)(CH2Ph) (10) and Dipp2NHSi(Br)(CH2Ph) (11). Reaction of Dipp2NHSi with the organoazides Ad-N3 (Ad=adamantyl) and TMS-N3 (TMS=trimethylsilyl) led to the formation of 1-Dipp2NHSi-2,5-bis(adamantyl)-tetrazoline (12) and bis(trimethylsilyl)amido azido silane (13), respectively. For 2,6-(diphenyl)phenyl-N3 C−H activation occurs and a cyclosilamine 14 was isolated.
N-Heterocyclic silylenes as ambiphilic activators and ligands
Mirjam J. Krahfuss and Udo Radius, Dalton Trans., 2021, 50, 6752.
This frontiers article highlights recent developments of the use of N-heterocyclic silylenes (NHSis), the higher homologues of the Arduengo-carbenes, as ambiphilic activators and ligands in organometallic chemistry and provides a comparison of five-membered ring NHSi ligands with ubiquitous N-heterocyclic carbene (NHC) and phosphine ligands. The frontier orbital region of NHSis differs considerably from that of NHCs which results in different ligation properties. The donor properties of NHSis are closer to those of phosphines than to those of NHCs. NHSis reveal a much stronger tendency to act as bridging ligands between two metal centres than NHCs or phosphines and NHSi insertion reactions into metal–ligand bonds are more facile to achieve compared to similar insertion reactions of NHCs. These interesting properties clearly distinguish NHSi ligands from their NHC or phosphine counterparts and should provide novel reactivities in basic organometallic chemistry and catalysis.
Cu-mediated vs. Cu-free selective borylation of aryl alkyl sulfones
Mingming Huang, Man Tang, Jiefeng Hu, Stephen A. Westcott, Udo Radius and Todd B. Marder, Chem. Commun., 2022, 58, 395.
A Cu-catalysed borylation of aryl alkyl sulfones was developed for the high yield synthesis of versatile arylboronic esters using a readily prepared NHC–Cu catalyst. In addition, the selective cleavage of either alkyl(C)–sulfonyl or aryl(C)–sulfonyl bonds of a cyclic sulfone via Cu-free or Cu-mediated processes generates the corresponding sulfinate salts, which can be further derivatised to provide sulfonyl-containing boronate esters, such as sulfones and sulfonyl fluorides.
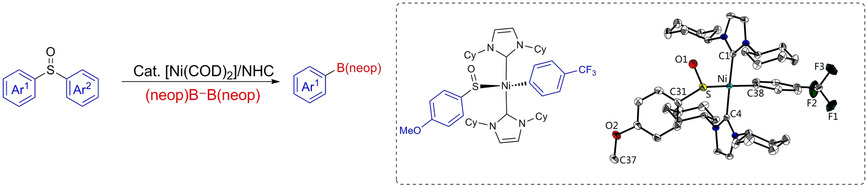
Ni-Catalyzed Borylation of Aryl Sulfoxides
Mingming Huang, Zhu Wu, Johannes Krebs, Alexandra Friedrich, Xiaoling Luo, Stephen A. Westcott, Udo Radius, and Todd B. Marder, Chem. Eur. J. 2021, 27, 8149–8158.
A nickel/N-heterocyclic carbene (NHC) catalytic system has been developed for the borylation of aryl sulfoxides with B2(neop)2 (neop=neopentyl glycolato). A wide range of aryl sulfoxides with different electronic and steric properties were converted into the corresponding arylboronic esters in good yields. The regioselective borylation of unsymmetric diaryl sulfoxides was also feasible leading to borylation of the sterically less encumbered aryl substituent. Competition experiments demonstrated that an electron-deficient aryl moiety reacts preferentially. The origin of the selectivity in the Ni-catalyzed borylation of electronically biased unsymmetrical diaryl sulfoxide lies in the oxidative addition step of the catalytic cycle, as oxidative addition of methoxyphenyl 4-(trifluoromethyl)phenyl sulfoxide to the Ni(0) complex occurs selectively to give the structurally characterized complex trans-[Ni(ICy)2(4-CF3-C6H4){(SO)-4-MeO-C6H4}] 4. For complex 5, the isomer trans-[Ni(ICy)2(C6H5)(OSC6H5)] 5-I was structurally characterized in which the phenyl sulfinyl ligand is bound via the oxygen atom to nickel. In solution, the complex trans-[Ni(ICy)2(C6H5)(OSC6H5)] 5-I is in equilibrium with the S-bonded isomer trans-[Ni(ICy)2(C6H5)(SOC6H5)] 5, as shown by NMR spectroscopy. DFT calculations reveal that these isomers are separated by a mere 0.3 kJ/mol (M06/def2-TZVP-level of theory) and connected via a transition state trans-[Ni(ICy)2(C6H5)(η2-{SO}-C6H5)], which lies only 10.8 kcal/mol above 5.
Ludwig Zapf, Udo Radius, and Maik Finze, Angew. Chem. Int. Ed. 2021, 60, 17974– 17980.
The 1,3-bis(tricyanoborane)imidazolate anion 1 was obtained in high yield from lithium imidazolate and B(CN)3−pyridine adduct. Anion 1 is chemically very robust and thus allowed the isolation of the corresponding H5O2+ salt. Furthermore, monoanion 1 served as starting species for the novel dianionic N-heterocyclic carbene (NHC), 1,3-bis(tricyanoborane)imidazoline-2-ylidenate anion 3 that acts as ditopic ligand via the carbene center and the cyano groups at boron. First reactions of this new NHC 3 with methyl iodide, elemental selenium, and [Ni(CO)4] led to the methylated imidazolate ion 4, the dianionic selenium adduct 5, and the dianionic nickel tricarbonyl complex 6. These NHC derivatives provide a first insight into the electronic and steric properties of the dianionic NHC 3. Especially the combination of properties, such as double negative charge, different coordination sites, large buried volume and good σ-donor and π-acceptor ability, make NHC 3 a unique and promising ligand and building block.
A General Synthetic Route to NHC-Phosphinidenes: NHC-mediated Dehydrogenation of Primary Phosphines
Luis Werner, Günther Horrer, Michael Philipp, Katharina Lubitz, Maximilian W. Kuntze-Fechner, and Udo Radius, Z. Anorg. Allg. Chem. 2021, 647, 881–8.
The dehydrocoupling of primary phosphines with N-heterocyclic carbenes (NHCs) to yield NHC-phosphinidenes is reported. The reaction of two equivalents of the NHCs Me2Im (1,3-dimethylimidazolin-2-ylidene), Me4Im (1,3,4,5-tetramethylimidazolin-2-ylidene), iPr2Im (1,3-di-iso-propylimidazolin-2-ylidene) and Mes2Im (2,4,6-trimethylphenylimidazolin-2-ylidene) with PhPH2 and MesPH2 led to the NHC stabilized phosphinidenes (NHC)PAr: (iPr2Im)PPh (1), (Mes2Im)PPh (2), (Me4Im)PPh (3), (Mes2Im)PMes (4), (Me2Im)PMes (5), (Me4Im)PMes (6) and (iPr2Im)PMes (7). The reaction of tBuPH2 with two equivalents of the NHCs afforded the corresponding NHC stabilized parent phosphinidenes (NHC)PH: (iPr2Im)PH (8), (Mes2Im)PH (9) and (Me4Im)PH (10). Reaction of 1 with oxygen and sulfur led to isolation of iPr2Im-P(O)2Ph (11) and iPr2Im-P(S)2Ph (12), whereas the reaction with elemental selenium and tellurium gave (NHC)PPh cleavage with formation of (iPr2Im)Se (13), iPr2ImTe (14) and different cyclo-oligophosphines. Furthermore, the complexes [{(iPr2Im)PPh}W(CO)5] (15), [Co(CO)2(NO){(iPr2Im)PPh}] (16) and [(η5-C5Me5)Co(η2-C2H4){(iPr2Im)PPh}] (17) have been prepared starting from 1 and a suitable transition metal complex precursor. The complexes 16 and 17 decompose in solution upon heating to ca. 80 °C to yield the NHC complexes [Co(iPr2Im)(CO)2(NO)] and [(η5-C5Me5)Co(iPr2Im)(η2-C2H4)] with formation of cyclo-oligophosphines. The reaction of 1 with [Ni(COD)2] afforded the diphosphene complex [Ni(iPr2Im)2(trans-PhP=PPh)] 18.
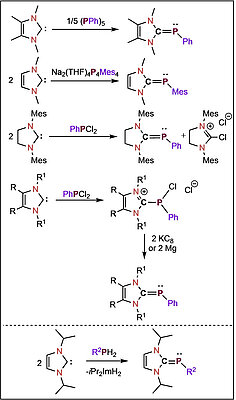
Lukas Tendera, Moritz Helm, Mirjam J. Krahfuss, Maximilian W. Kuntze-Fechner, and Udo Radius, Chem. Eur. J. 2021, 27, 17849–17861.
A case study on the effect of the employment of two different NHC ligands in complexes [Ni(NHC)2] (NHC=iPr2ImMe 1Me, Mes2Im 2) and their behavior towards alkynes is reported. The reaction of a mixture of [Ni2(iPr2ImMe)4(μ-(η2 : η2)-COD)] B/ [Ni(iPr2ImMe)2(η4-COD)] B’ or [Ni(Mes2Im)2] 2, respectively, with alkynes afforded complexes [Ni(NHC)2(η2-alkyne)] (NHC=iPr2ImMe: alkyne=MeC≡CMe 3, H7C3C≡CC3H7 4, PhC≡CPh 5, MeOOCC≡CCOOMe 6, Me3SiC≡CSiMe3 7, PhC≡CMe 8, HC≡CC3H7 9, HC≡CPh 10, HC≡C(p-Tol) 11, HC≡C(4-tBu-C6H4) 12, HC≡CCOOMe 13; NHC=Mes2Im: alkyne=MeC≡CMe 14, MeOOCC≡CCOOMe 15, PhC≡CMe 16, HC≡C(4-tBu-C6H4) 17, HC≡CCOOMe 18). Unusual rearrangement products 11 a and 12 a were identified for the complexes of the terminal alkynes HC≡C(p-Tol) and HC≡C(4-tBu-C6H4), 11 and 12, which were formed by addition of a C−H bond of one of the NHC N-iPr methyl groups to the C≡C triple bond of the coordinated alkyne. Complex 2 catalyzes the cyclotrimerization of 2-butyne, 4-octyne, diphenylacetylene, dimethyl acetylendicarboxylate, 1-pentyne, phenylacetylene and methyl propiolate at ambient conditions, whereas 1Me is not a good catalyst. The reaction of 2 with 2-butyne was monitored in some detail, which led to a mechanistic proposal for the cyclotrimerization at [Ni(NHC)2]. DFT calculations reveal that the differences between 1Me and 2 for alkyne cyclotrimerization lie in the energy profile of the initiation steps, which is very shallow for 2, and each step is associated with only a moderate energy change. The higher stability of 3 compared to 14 is attributed to a better electron transfer from the NHC to the metal to the alkyne ligand for the N-alkyl substituted NHC, to enhanced Ni-alkyne backbonding due to a smaller CNHC−Ni−CNHC bite angle, and to less steric repulsion of the smaller NHC iPr2ImMe.
First-Row d‑Block Element-Catalyzed Carbon−Boron Bond Formation and Related Processes
Shubhankar Kumar Bose, Lujia Mao, Laura Kuehn, Udo Radius, Jan Nekvinda, Webster L. Santos, Stephen A. Westcott, Patrick G. Steel, and Todd B. Marder, Chem. Rev. 2021, 121, 21, 13238–13341.
Organoboron reagents represent a unique class of compounds because of their utility in modern synthetic organic chemistry, often affording unprecedented reactivity. The transformation of the carbon−boron bond into a carbon−X (X = C, N, and O) bond in a stereocontrolled fashion has become invaluable in medicinal chemistry, agrochemistry, and natural products chemistry as well as materials science. Over the past decade, first-row d-block transition metals have become increasingly widely used as catalysts for the formation of a carbon−boron bond, a transformation traditionally catalyzed by expensive precious metals. This recent focus on alternative transition metals has enabled growth in fundamental methods in organoboron chemistry. This review surveys the current state-of-the-art in the use of first-row d-block element-based catalysts for the formation of carbon−boron bonds.
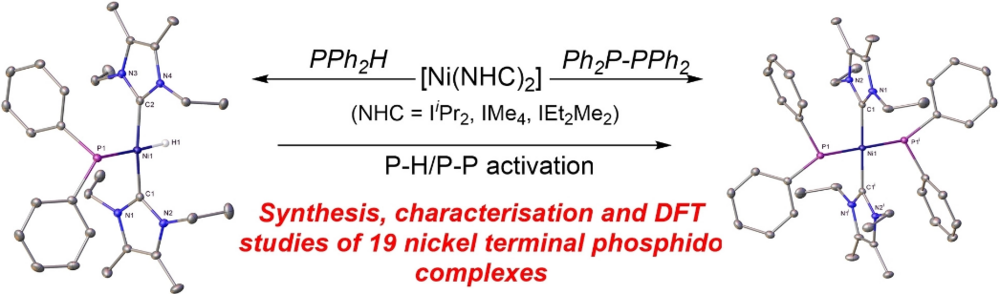
Sara Sabater, David Schmidt, Heidi (née Schneider) Schmidt, Maximilian W. Kuntze-Fechner, Thomas Zell, Connie J. Isaac, Nasir A. Rajabi, Harry Grieve, William J. M. Blackaby, John P. Lowe, Stuart A. Macgregor, Mary F. Mahon, Udo Radius, and Michael K. Whittlesey, Chem. Eur. J. 2021, 27, 13221–13234.
The addition of PPh2H, PPhMeH, PPhH2, P(para-Tol)H2, PMesH2 and PH3 to the two-coordinate Ni0 N-heterocyclic carbene species [Ni(NHC)2] (NHC=IiPr2, IMe4, IEt2Me2) affords a series of mononuclear, terminal phosphido nickel complexes. Structural characterisation of nine of these compounds shows that they have unusual trans [H−Ni−PR2] or novel trans [R2P−Ni−PR2] geometries. The bis-phosphido complexes are more accessible when smaller NHCs (IMe4>IEt2Me2>IiPr2) and phosphines are employed. P−P activation of the diphosphines R2P−PR2 (R2=Ph2, PhMe) provides an alternative route to some of the [Ni(NHC)2(PR2)2] complexes. DFT calculations capture these trends with P−H bond activation proceeding from unconventional phosphine adducts in which the H substituent bridges the Ni−P bond. P−P bond activation from [Ni(NHC)2(Ph2P−PPh2)] adducts proceeds with computed barriers below 10 kcal mol−1. The ability of the [Ni(NHC)2] moiety to afford isolable terminal phosphido products reflects the stability of the Ni−NHC bond that prevents ligand dissociation and onward reaction.
C–F Bond Activation of perfluorinated Arenes using NHC-stabilized Cobalt Half-Sandwich Complexes
Daniel Ertler, Maximilian W. Kuntze-Fechner, Simon Dürr, Katharina Lubitz and Udo Radius, New J. Chem., 2021, 45, 14999
A study on the reactivity of NHC cobalt half-sandwich complexes [(η5-C5R15)Co(R22Im)(η2-C2H4)] (R1 = H; R2 = Me 1, iPr 2) and [(η5-C5R15)Co(R22Im)(η2-C2H3{SiMe3})] (R1 = H, R2 = Me 3, iPr 4; R1 = Me, R2 = Me 5, iPr 6) towards selected perfluoroarenes is presented (R2Im = 1,3-di(organyl)-imidazolin-2-ylidene). The reaction with hexafluorobenzene, perfluorotoluene and decafluorobiphenyl at 100 °C led to the isolation of the cobalt(II) complexes [CpCo(iPr2Im)(C6F5)] 7, [CpCo(iPr2Im)(C7F7)] 8, [CpCo(iPr2Im)(C12F9)] 9, [Cp*Co(iPr2Im)(C6F5)] 10, [Cp*Co(iPr2Im)(C7F7)] 11 and [Cp*Co(iPr2Im)(C12F9)] 12 (Cp* = η5-C5{CH3}5), Cp = η5-C5H5). The cobalt(II) fluoride [CpCo(iPr2Im)(F)] was detected as a side product of these reactions. The reaction of [CpCo(R22Im)(η2-C2H4)] (R2 = Me 1, iPr 2) with C6F6 and C7F8 at 60 °C afforded the dinuclear complexes [{CpCo(R22Im)}2(μ-η2,η2-C6F6)] (R2 = Me 13, iPr 14) and [{CpCo(R22Im)}2(μ-η2,η2-C7F8)] (R2 = Me 15, iPr 16). Furthermore, the dinuclear complexes [{CpCo(Me2Im)}2(μ-η2,η2-C10F8)] 17, [{CpCo(iPr2Im)}2(μ-η2,η2-C10F8)] 18 and mononuclear [CpCo(iPr2Im)(η2-C10F8)] 19 were isolated from the reaction of 1 and 2 with octafluoronaphthalene at room temperature. Based on the experimental data a mechanism is proposed for the C–F bond activation of perfluoroarenes with complexes [Cp(*)Co(NHC)(olefin)]. Transfer of [Cp(*)Co(NHC)] to the perfluoroarene, which is limited by the activation barrier to replace the alkene ligand, affords mononuclear cobalt complexes [Cp(*)Co(NHC)(η2-ArF)]. Ligand dismutation at higher temperatures leads to dinuclear complexes [{Cp(*)Co(NHC)}2(μ-η2,η2-ArF)], which are precursors for the formation of the cobalt(II) complexes [Cp(*)Co(NHC)(ArF)] and [Cp(*)Co(NHC)(F)]. One electron oxidative addition prevails in the C–F bond activation step using these cobalt half-sandwich complexes.
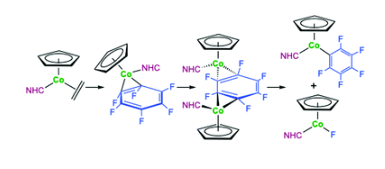
N-Heterocyclic Carbene and Cyclic (Alkyl)(amino)carbene Adducts of Antimony(III)
M. S. M. Philipp, M. J. Krahfuss, K. Radacki, U. Radius, Eur. J. Inorg. Chem., 2021.
A systematic study on Lewis-acid/base adducts of Nheterocyclic carbenes (NHCs) and the cyclic (alkyl)(amino)carbene cAACMe (1-(2,6-di-iso-propylphenyl)-3,3,5,5-tetramethyl-pyrrolidin-2ylidene) with antimony(III) chlorides of the general formula SbCl2R (R = Cl, Ph, Mes) is presented. The reaction of the NHCs Me2ImMe (1,3,4,5-tetra-methyl-imidazolin-2-ylidene), iPr2ImMe (1,3-di-isopropyl4,5-dimethyl-imidazolin-2-ylidene), Mes2Im, Dipp2Im (R2Im = 1,3-diorganyl-imidazolin-2-ylidene; Mes = 2,4,6-trimethylphenyl, Dipp = 2,6di-isopropylphenyl) and cAACMe with antimony(III) compounds SbCl2R (R = Cl (1), Ph (2) and Mes (3)) yields the adducts NHC∙SbCl2R (R = Cl (4), Ph (5) and Mes (6); NHC = Me2ImMe (a), iPr2ImMe (b), Dipp2Im (c) and Mes2Im (d)) and cAACMe∙SbCl2R (R = Cl (4e) and Ph (5e)). Thermal treatment of (Dipp2Im)∙SbCl2Ar (Ar = Ph (5c) and Mes (6c)) in benzene leads to isomerization to the backbone coordinated aNHC-adduct aDipp2Im∙SbCl2Ar (Ar = Mes (7) and Ph (8)) (“a” denotes “abnormal” coordination mode of the NHC) in high yields. One of the chloride substituents at antimony of 7 can be abstracted by GaCl3 or Ag[BF4] to obtain the imidazolium salts [aDipp2Im∙SbClMes][BF4] (9) and [aDipp2Im∙SbClMes][GaCl4] (10).
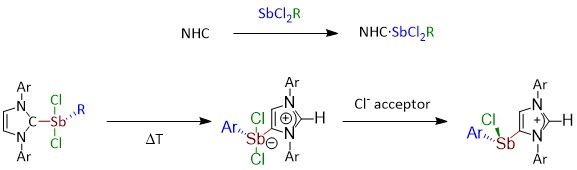
S. A. Föhrenbacher, M. J. Krahfuß, L. Zapf, A. Friedrich, N. Ignat’ev, M. Finze, U. Radius, Chem. Eur. J. 2021, 27, 3504-3516.
Fluoride abstraction from different types of transition metal fluorido complexes [LnM−F] using the Lewis acid tris(pentafluoroethyl)difluorophosphorane (C2F5)3PF2 to yield cationic transition metal complexes with the tris(pentafluoroethyl)trifluorophosphate counterion (FAP anion, [(C2F5)3PF3]−) is reported. (C2F5)3PF2 reacted with trans-[Ni(iPr2Im)2(F)(ArF)] (iPr2Im = 1,3-di(iso-propyl)-imidazolin-2-ylidene; ArF = C6F5 1a; 4−CF3-C6F4 1b; 4−C6F5-C6F4 1c) via fluoride transfer to form the complex salts trans-[Ni(iPr2Im)2(solv)(ArF)]FAP (2a–c[solv]) (solv = Et2O, CH2Cl2, THF) depending on the reaction medium. In the presence of stronger Lewis bases such as carbenes or PPh3, solvent coordination was suppressed and the complexes trans-[Ni(iPr2Im)2(PPh3)(C6F5)]FAP (trans-2a[PPh3]) and cis-[Ni(iPr2Im)2(Dipp2Im)(C6F5)]FAP (cis-2a[Dipp2Im]) (Dipp2Im = 1,3-bis(2,6-di-iso-propylphenyl)-imidazolin-2-ylidene) were isolated. Fluoride abstraction from [(Dipp2Im)CuF] (3) in CH2Cl2 or 1,2-difluorobenzene led to the isolation of [{(Dipp2Im)Cu}2]2+2FAP- (4). Subsequent reaction of 4 with PPh3 and different carbenes resulted in the complexes [(Dipp2Im)Cu(LB)]FAP (LB = Lewis base) (5a–e). In the presence of C6Me6, fluoride transfer afforded [(Dipp2Im)Cu(C6Me6)]FAP (5f). The reactivity of 4 demonstrates that it serves as a source of cationic [(Dipp2Im)Cu)]+. Fluoride-ion abstraction of Cp2TiF2 (7) resulted in the formation of dinuclear [(F)(Cp)2Ti(μ-F)Ti(Cp)2(F)]FAP (8) with one terminal fluorido ligand at each titanium atom and an additional bridging fluorido ligand.
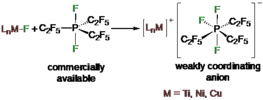
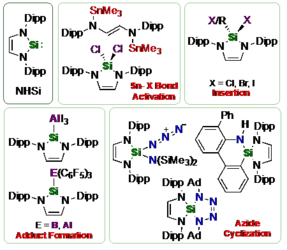
M. J. Krahfuss, U. Radius, Eur. J. Inorg. Chem. 2021, 548-561.
Investigations concerning the reactivity of the N-heterocyclic silylene Dipp2NHSi (1, 1,3-bis(2,6-diisopropylphenyl)-1,3-diaza-2-silacyclopent-4-en-2-ylidene) towards selected alanes and boranes, elemental halides X2 (X = Br, I), selected halide containing substrates such as tin chlorides and halocarbons, as well as organoazides are presented. The NHSi adducts Dipp2NHSi·AlI3 (2), Dipp2NHSi·Al(C6F5)3 (3) and Dipp2NHSi·B(C6F5)3 (4) were formed by reaction of Dipp2NHSi with the corresponding Lewis acids AlI3, Al(C6F5)3 and B(C6F5)3. Adducts 3 and 4 were tested with respect to their ability to activate small organic molecules, but no frustrated Lewis pair reactivity was observed. Reactions of Dipp2NHSi with Br2, I2, Ph2SnCl2 and Me3SnCl led to formation of Dipp2NHSiBr2 (5), Dipp2NHSiI2 (6), Dipp2NHSiCl2 (7) and {(Me3Sn)N(Dipp)CH}2 (8), respectively. The reaction with the halocarbons methyl iodide, benzyl chloride and benzyl bromide afforded the insertion products Dipp2NHSi(I)(CH3) (9), Dipp2NHSi(Cl)(CH2Ph) (10) and Dipp2NHSi(Br)(CH2Ph) (11). Reaction of Dipp2NHSi with the organoazides Ad-N3 (Ad = adamantyl) and TMS-N3 (TMS = trimethylsilyl) led to the formation of 1-Dipp2NHSi-2,5-bis(adamantyl)-tetrazoline (12) and bis(trimethylsilyl)amido azido silane (13), respectively. For 2,6-(diphenyl)phenyl-N3 C–H activation occurs and a cyclosilamine 14 was isolated.
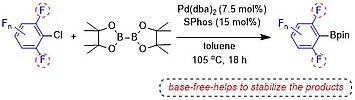
Base-Free Pd-catalyzed C-Cl Borylation of Fluorinated Aryl Chlorides
Y. P. Budiman, S. Lorenzen, Z. Liu, U. Radius, T. B. Marder, Chem. Eur. J. 2021, 27, 3869-3874.
Catalytic C–X borylation of aryl halides containing two ortho-fluorines has been found to be challenging, as most previous methods require stoichiometric amounts of base and the polyfluorinated aryl boronates suffer from protodeboronation, which is accelerated by ortho-fluorine substituents. Herein, we report that a combination of Pd(dba)2 (dba = dibenzylideneacetone) with SPhos (2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl) as a ligand is efficient to catalyze the C–Cl borylation of aryl chlorides containing two ortho-fluorine substituents. This method, conducted under base-free conditions, is compatible with the resulting di-ortho-fluorinated aryl boronate products which are sensitive to base.
Z. Liu, Y. P. Budiman, Y. Tian, A. Friedrich, M. Huang, S. A. Westcott, U. Radius, T. B. Marder, Chem. Eur. J. 2020, 26, 17267-17274.
Boron Helps Make Fluorinated Alkynes: We reported a mild procedure for the copper-catalyzed oxidative cross-coupling of electron-deficient polyfluorophenylboronate esters with terminal alkynes. This method displays good functional group tolerance and broad substrate scope, generating cross-coupled alkynyl(fluoro)arene products in moderate to excellent yields. Thus, it represents a simple and alternative to the conventional Sonogashira reaction.
Bis-NHC Aluminium and Gallium dihydride cations [(NHC)2EH2]+ (E = Al, Ga)
A. Hock, L. Werner, M. Riethmann, U. Radius, Eur. J. Inorg. Chem. 2020, 4015-4023.
The NHC alane and gallane adducts (NHC)∙AlH2I (NHC = Me2ImMe 7, iPr2Im 8, iPr2ImMe 9) and (NHC)∙GaH2I (NHC = Me2ImMe 10, iPr2ImMe 11, Dipp2Im 12; R2Im = 1,3‐di-organyl-imidazolin-2-ylidene; Dipp = 2,6 diisopropylphenyl; iPr = isopropyl; Me2ImMe = 1,3,4,5-tetra-methyl-imidazolin-2-ylidene) were prepared either by the simple yet efficient reaction of the NHC adduct (NHC)∙AlH3 with elemental iodine or by the treatment of (NHC)∙GaH3 with an excess of methyl iodide at room temperature. The reaction of one equivalent of the group 13 NHC complexes with an additional equivalent of the corresponding NHC afforded cationic aluminium and gallium hydrides [(NHC)2∙AlH2]I (NHC = Me2ImMe 13, iPr2Im 14, iPr2ImMe 15) and [(NHC)2∙GaH2]I (NHC = Me2ImMe 16, iPr2ImMe 17) and the normal and abnormal NHC coordinated compound [(Dipp2Im)∙GaH2(aDipp2Im)]I 18. Compounds 7-18 were isolated and characterized by means of elemental analysis, IR and multinuclear NMR spectroscopy and by X-ray diffraction of the compounds 7, 9, 10, 15, 16 and 18.
M. W. Kuntze-Fechner, H. Verplancke, L. Tendera, M. Diefenbach, I. Krummenacher, H. Braunschweig, T. B. Marder, M. C. Holthausen, U. Radius, Chem. Sci. 2020, 11, 11009-11023.
(2020 Chemical Science HOT Article Collection)
The reaction of [Ni(Mes2Im)2] (1) (Mes2Im = 1,3-dimesityl-imidazolin-2-ylidene) with polyfluorinated arenes as well as mechanistic investigations concerning the insertion of 1 and [Ni(iPr2Im)2] (1ipr) (iPr2Im = 1,3‐diisopropyl-imidazolin-2-ylidene) into the C–F bond of C6F6 is reported. The reaction of 1 with different fluoroaromatics leads to formation of the nickel fluoroaryl fluoride complexes trans-[Ni(Mes2Im)2(F)(ArF)] (ArF = 4‑CF3‑C6F4 2, C6F5 3, 2,3,5,6‑C6F4N 4, 2,3,5,6‑C6F4H 5, 2,3,5‑C6F3H2 6, 3,5‑C6F2H3 7) in fair to good yields with the exception of the formation of the pentafluorophenyl complex 3 (less than 20 %). Radical species and other diamagnetic side products were detected for the reaction of 1 with C6F6, in line with a radical pathway for the C–F bond activation step using 1. The difluoride complex trans-[Ni(Mes2Im)2(F)2] (9), the bis(aryl) complex trans-[Ni(Mes2Im)2(C6F5)2] (15), the structurally characterized nickel(I) complex trans-[NiI(Mes2Im)2(C6F5)] (11) and the metal radical trans‑[NiI(Mes2Im)2(F)] (12) were identified. Complex 11, and related [NiI(Mes2Im)2(2,3,5,6‑C6F4H)] (13) and [NiI(Mes2Im)2(2,3,5-C6F3H2)] (14), were synthesized independently by reaction of trans-[Ni(Mes2Im)2(F)(ArF)] with PhSiH3. Simple electron transfer from 1 to C6F6 was excluded, as the redox potential of the reaction partner does not match and [Ni(Mes2Im)2]+, which was prepared independently, was not detected. DFT calculations were performed on the insertion of [Ni(iPr2Im)2] (1ipr) and [Ni(Mes2Im)2] (1) into the C–F bond of C6F6. For 1ipr, a concerted and an NHC-assisted pathway were identified as pathways with the lowest kinetic barriers, whereas for 1, a radical mechanism with fluoride abstraction and an NHC-assisted pathway are both associated with almost the same kinetic barrier.

Visible-Light-Induced Ni-Catalyzed Radical Borylation of Chloroarenes
Y. Tian, X. Guo, I. Krummenacher, Z. Wu, J. Nitsch, H. Braunschweig, U. Radius, T. B. Marder, J. Am. Chem. Soc. 2020, 142, 18231-18242.
A highly selective and general photo-induced C-Cl borylation protocol that employs [Ni(IMes)2] (IMes = 1,3-dimesitylimidazoline-2-ylidene) for the radical borylation of chloroarenes is reported. This photo-induced system operates with visible light (400 nm) and achieves borylation of a wide range of chloroarenes with B2pin2 at room temperature in excellent yields and with high selectivity, thereby demonstrating its broad utility and functional group tolerance. Mechanistic investigations suggest that the borylation reactions proceed via a radical process. EPR studies demonstrate that [Ni(IMes)2] undergoes very fast chlorine atom abstraction from aryl chlorides to give [NiⅠ(IMes)2Cl] and aryl radicals. Control experiments indicate that light promotes the reaction of [NiⅠ(IMes)2Cl] with aryl chlorides generating additional aryl radicals and [NiII(IMes)2Cl2]. The aryl radicals react with an anionic sp2-sp3 diborane [B2pin2(OMe)]- formed from B2pin2 and KOMe to yield the corresponding borylation product and the [Bpin(OMe)]•- radical anion, which reduces [NiII(IMes)2Cl2] under irradiation to regenerate [NiⅠ(IMes)2Cl] and [Ni(IMes)2] for the next catalytic cycle.

A. Hock, L. Werner, C. Luz, U. Radius, Dalton Trans. 2020, 49, 11108-11119.
The synthesis and characterization of N-Heterocyclic carbene (NHC) and cyclic (alkyl)(amino)carbene (cAAC) gallane and chlorogallane adducts of the type (NHC)∙GaH3 (NHC = Me2ImMe 1, iPr2Im 2 and Dipp2ImH 3; Me2ImMe = 1,3,4,5-tetra-methyl-imidazolin-2-ylidene; R2Im = 1,3‐di-organyl-imidazolin-2-ylidene; Dipp = 2,6-diisopropylphenyl; Dipp2ImH= 1,3‐bis(2,6-diisopropylphenyl)-imidazolidin-2-ylidene), (NHC)∙GaH2Cl (NHC = iPr2ImMe 9, Dipp2Im 10 and Dipp2ImH 11; iPr2ImMe = 1,3 diisopropyl-4,5-dimethyl-imidazolin-2-ylidene), (NHC)∙GaHCl2 (NHC = iPr2ImMe 12, Dipp2Im 13 and Dipp2ImH 14) and (cAACMe)∙GaHCl2 15 is reported. Compounds 1-3 and 9-11 are unstable in solution as heating to the boiling of toluene (110°C) leads to decomposition into elemental gallium and the corresponding dihydroaminal NHC-H2. Reaction of the mono-NHC adducts with a second equivalent of NHC also afforded decomposition and formation of NHC-H2, whereas the reaction of the NHC-stabilized gallanes with one equivalent cAACMe leads to insertion of the cAACMe carbene carbon atom into the Ga-H bond. The synthesis and characterization of (NHC)∙GaH2(cAACMeH) (NHC = Me2ImMe 5, iPr2ImMe 6 and Dipp2Im 7), the products of an oxidative addition of (NHC)∙GaH3 to cAACMe, are presented. The adduct (Dipp2ImH)∙GaH3 3 reacts with two equivalents of cAACMe under release of Dipp2ImH and insertion of two cAACMe molecules into the Ga-H bonds to give the bisalkylgallane (cAACMeH)2GaH 8. If one or two hydrogen atoms of the NHC gallane adducts are replaced with an electronic withdrawing chloride substituent selective insertion of the cAACMe into the Ga-H bond occurs only for the adducts of sterically less demanding NHCs such as iPr2ImMe. The reaction of cAACMe with (iPr2ImMe)∙GaH2Cl 9 and (iPr2ImMe)∙GaHCl2 12 afforded (iPr2ImMe)∙GaHCl(cAACMeH) 16 and (iPr2ImMe)∙GaCl2(cAACMeH) 17. The reaction of cAACMe with (NHC)∙GaHCl2 and (NHC)∙GaH2Cl (NHC = Dipp2Im, Dipp2ImH), adducts of the sterically more demanding NHCs, lead with extrusion of the NHC to (cAACMe)∙GaHCl2 15 and (cAACMeH)2GaCl 18.
Ni-Catalyzed Traceless, Directed C3-Selective C-H Borylation of Indoles
Y. Tian, X. Guo, Z. Wu, A. Friedrich, S. A. Westcott, H. Braunschweig, U. Radius, T. B. Marder, J. Am. Chem. Soc. 2020, 142, 13136-13144.
A highly efficient and general protocol for traceless, directed C3-selective C-H borylation of indoles with [Ni(IMes)2] as the catalyst is reported. Activation and borylation of N-H bonds by [Ni(IMes)2] is essential to install a Bpin moiety at the N-position as a traceless directing group, which enables the C3-selective borylation of C-H bonds. The N-Bpin group which is formed is easily converted in situ back to an N-H group by the oxidiative addition product of [Ni(IMes)2] and in situ-generated HBpin. The catalytic reactions are operationally simple, allowing borylation of a variety of substituted indoles with B2pin2 in excellent yields and with high selectivity. The C-H borylation can be followed by Suzuki-Miyaura cross-coupling of the C-borylated indoles in an overall two-step, one-pot process providing an efficient method for synthesizing C3-functionalized heteroarenes.
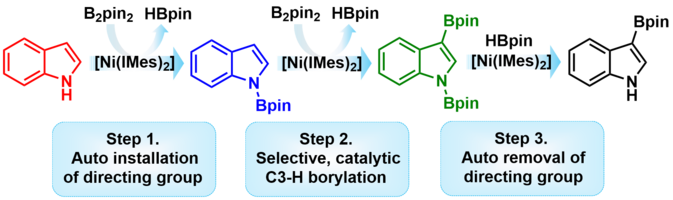
L. Tendera, T. Schaub, M. J. Krahfuss, M. W. Kuntze-Fechner, U. Radius, Eur. J. Inorg. Chem. 2020, 3194-3207.
Investigations concerning the reactivity of Ni(0) complexes [Ni(NHC)2] of NHCs (N-heterocyclic carbene) of different steric demand, i.e. Mes2Im (= 1,3-dimesitylimidazolin-2-ylidene) and iPr2Im (= 1,3‐di-iso-propyl-imidazolin-2-ylidene), with olefins, ketones and aldehydes are reported. The reaction of [Ni(Mes2Im)2] 1 with ethylene or methyl acrylate afforded the complexes [Ni(Mes2Im)2(η2-C2H4)] 3 and [Ni(Mes2Im)2(η2‑(C,C)‑H2C=CHCOOMe)] 4, as it was previously reported for [Ni2(iPr2Im)4(µ‑(η2:η2)‑COD)] 2 as a source for [Ni(iPr2Im)2]. In contrast to 2, complex 1 does not react with sterically more demanding olefins such as tetramethylethylene, 1,1-diphenylethylene and cyclohexene. The reaction of [Ni(NHC)2] with more π-acidic ketones or aldehydes led to formation of complexes with side-on η2-(C,O)‑coordinating ligands, i.e. [Ni(iPr2Im)2(η2-O=CHtBu)] 5, [Ni(iPr2Im)2(η2-O=CHPh)] 6, [Ni(iPr2Im)2(η2-O=CMePh)] 7, [Ni(iPr2Im)2(η2-O=CPh2)] 8, [Ni(iPr2Im)2(η2-O=C(4-F-C6H4)2)] 9, [Ni(iPr2Im)2(η2-O=C(OMe)(CF3))] 10 and [Ni(Mes2Im)2(η2-O=CHPh)] 11, [Ni(Mes2Im)2(η2-O=CH(CH(CH3)2))] 12, [Ni(Mes2Im)2(η2-O=CH(4-NMe2-C6H4))] 13, [Ni(Mes2Im)2(η2-O=CH(4-OMe-C6H4))] 14, [Ni(Mes2Im)2(η2-O=CPh2)] 15 and [Ni(Mes2Im)2(η2-O=C(4-F-C6H4)2)] 16. The reaction of 1 and 2 with these simple aldehydes and ketones does not lead to a significantly different outcome, but NHC ligand rotation is hindered for the Mes2Im complexes 3, 4 and 11-16 according to NMR spectroscopy. The solid state structures of 3, 4, 11 and 12 reveal significantly larger CNHC-Ni-CNHC angles in the Mes2Im complexes compared to the iPr2Im complexes. As electron transfer in d8-( or d10-) ML2 complexes to π-acidic ligands depends on the L-M-L bite angle, the different NHCs lead thus to a different degree of electron transfer and activation of the olefin, aldehyde or ketone ligand, i.e. [Ni(iPr2Im)2] is the better donor to these π-acidic ligands. Furthermore, we identified two different side products from the reaction of 1 with benzaldehyde, i.e. trans-[Ni(Mes2Im)2H(OOCPh)] 17 and [Ni2(Mes2Im)2(µ2‑CO)(µ2‑η2‑C,O‑PhCOCOPh)] 18, which indicate that radical intermediates and electron transfer processes might be of importance in the reaction of 1 with aldehydes and ketones.
M. J. Krahfuss, J. Nitsch, F. M. Bickelhaupt, T. B. Marder, Udo Radius, Chem. Eur. J. 2020, 26, 11276-11292.
A study on the reactivity of the N-heterocyclic silylene Dipp2NHSi (1,3-bis(diisopropylphenyl)-1,3-diaza-2-silycyclopent-4-en-2-yliden) with the transition metal complexes [Ni(CO)4], [M(CO)6] (M= Cr, Mo, W), [Mn(CO)5(Br)] and [(η5‑C5H5)Fe(CO)2(I)] is reported. We demonstrate that N-heterocyclic silylenes, the higher homologues of the now ubiquitous NHC ligands, show a remarkably different behavior in coordination chemistry compared to NHC ligands. Calculations on the electronic features of these ligands revealed significant differences in the frontier orbital region which lead to some peculiarities of the coordination chemistry of silylenes, as demonstrated by the synthesis of the dinuclear, NHSi-bridged complex [{Ni(CO)2(μ-Dipp2NHSi)}2] (2), complexes [M(CO)5(Dipp2NHSi)] (M = Cr 3, Mo 4, W 5), [Mn(CO)3(Dipp2NHSi)2(Br)] (9) and [(η5‑C5H5)Fe(CO)2(Dipp2NHSi-I)] (10). DFT calculations on several model systems [Ni(L)], [Ni(CO)3(L)], and [W(CO)5(L)] (L = NHC, NHSi) reveal that carbenes are typically the much better donor ligands with a larger intrinsic strength of the metal-ligand bond. The decrease going from the carbene to the silylene ligand is mainly caused by favorable electrostatic contributions for the NHC ligand to the total bond strength, whereas the orbital interactions were often found to be higher for the silylene complexes. Furthermore, we have demonstrated that the contribution of σ- and π-interaction depends significantly on the system under investigation. The σ-interaction is often much weaker for the NHSi ligand compared to NHC but, interestingly, the π-interaction prevails for many NHSi complexes. For the carbonyl complexes, the NHSi ligand is the better σ-donor ligand, and contributions of π-symmetry play only a minor role for the NHC and NHSi co-ligands.
N-Heterocyclic Silylenes as Metal-Metal and Metal-Halide Activators in Transition Metal Complexes
M. J. Krahfuss, U. Radius, Inorg. Chem. 2020, 59, 10976-10985.
Several examples are provided that NHSi (N-heterocyclic silylene) ligands are much better metal metal bridging ligands as compared to NHCs and that NHSis also tend to insert much more easily into metal halide bonds of transition metal complexes. Dipp2NHSi (1,3-bis(2,6-diisopropyphenyl)-1,3-diaza-4-silacyclopent-4-en-2-yliden) reacts with {[Fe(CO)2(η5-C5H5)]2} and [(η5-C5H5)2Ni] to yield, in contrast to the results obtained for analogous transformations with NHCs, the dinuclear silylene-bridged complexes [Fe2(CO)2(µ-CO)(η5-C5H5)(µ-Dipp2NHSi)] 1 and [(η5-C5H5)Ni(µ-Dipp2NHSi)]2 2. The reaction with [(η5-C5H5)Ni(PPh3)(Cl)] leads with insertion of the silylene into the nickel–chloride bond to [(η5-C5H5)Ni(PPh3)(Dipp2NHSi-Cl)] 3. [Mn(CO)5(Cl)] reacts with two equivalents Dipp2NHSi to afford [Mn(CO)4(Dipp2NHSi)(Dipp2NHSi-Cl)] 5, in which one of the silylenes has inserted into the Mn–Cl bond and acts as a chloro silyl ligand. The reaction of two equivalents Dipp2NHSi with the corresponding iodide complex [Mn(CO)5(I)] leads to formation of the bis-silylene complex [Mn(CO)3(Dipp2NHSi)2(I)] 4.
Y. P. Budiman, A. Jayaraman, A. Friedrich, F. Kerner, U. Radius, T. B. Marder, J. Am. Chem. Soc. 2020, 142, 6036–6050.
Abstract
C–C reductive elimination from [PdL2(C6F5)2] to form polyfluorinated biaryls has been a challenge for over 50 years. Thus, palladium-catalyzed homocoupling of arylboronates (ArF–Bpin) containing two ortho-fluorine substituents is very difficult as the reaction typically stops at the [PdL2(ArF)2] stage after two transmetalation steps. The transmetalated complexes cis-[Pd(MeCN)2(C6F5)2] (3a), cis-[Pd(MeCN)2(2,4,6-C6F3H2)2] (3b), and cis-[Pd(MeCN)2(2,6-C6F2H3)2] (3e) have been isolated from the reaction of ArF–Bpin with Pd(OAc)2 in acetonitrile solvent, with no homocoupling observed. However, catalytic homocoupling proceeds smoothly in a “weakly-coordinating” arene solvent as long as no ancillary ligands or coordination solvents are present. DFT computations performed reveal that the active catalyst formed by arene solvent coordination leads to an overall reduced barrier for the reductive elimination step compared to the formation of stable [PdL2(ArF)2] complexes in the presence of a donor ligand or solvent L.
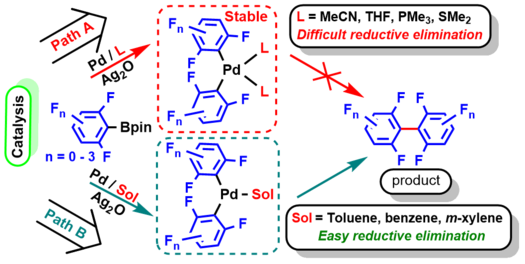
J. H. J. Berthel, M. J. Krahfuß, U. Radius, Z. Anorg. Allg. Chem. 2020, 646, 692-704.
Abstract
A novel, useful in situ synthesis for NHC nickel allyl halide complexes [Ni(NHC)(η3-allyl)(X)] starting from [Ni(CO)4], NHC and allyl halides is presented. The reaction of [Ni(CO)4] with (i) one equivalent of the corresponding NHC and (ii) with an excess of the corresponding allyl chloride at room temperature leads with elimination of carbon monoxide to complexes of the type [Ni(NHC)(η3-allyl)(X)]. This approach has been used to synthesize the complexes [Ni(tBu2Im)(η3-H2C-C(Me)-CH2)(Cl)] 2, [Ni(iPr2ImMe)(η3-H2C-C(Me)-CH2)(Cl)] 3, [Ni(iPr2Im)(η3-H2C-C(Me)-CH2)(Cl)] 4, [Ni(iPr2Im)(η3-H2C-C(H)-C(Me)2)(Br)] 5, [Ni(Me2ImMe)(η3-H2C-C(Me)-CH2)(Cl)] 6 and [Ni(EtiPrImMe)(η3-H2C-C(Me)-CH2)(Cl)] 7. The complexes 1 to 7 were characterized using NMR and IR spectroscopy and elemental analysis, and the molecular structures are provided for 2 and 7. The allyl nickel complexes 1-7 are stereochemically non-rigid in solution due to (i) NHC rotation about the nickel-carbon bond, (ii) allyl rotation about the Ni-η3-allyl axis and (iii) π-σ-π allyl isomerization processes. The allyl halide complexes can be methylated as has been demonstrated by the methylation of a number of the complexes [Ni(NHC)(η3-allyl)(X)] with methyl magnesium chloride or methyl lithium, which led to isolation of the complexes [Ni(Me2Im)(η3-H2C-C(Me)-CH2)(Me)] 8, [Ni(tBu2Im)(η3-H2C-C(Me)-CH2)(Me)] 9, [Ni(iPr2ImMe)(η3-H2C-C(Me)-CH2)(Me)] 10, [Ni(iPr2Im)(η3-H2C-C(Me)-CH2)(Me)] 11, [Ni(iPr2Im)(η3-H2C-C(H)-C(Me)2)(Me)] 12, and [Ni(EtiPrImMe)(η3-H2C-C(Me)-CH2)(Me)] 13. These complexes have been fully characterized including X-ray molecular structures for 10 and 11.
G. Horrer, M. J. Krahfuß, K. Lubitz, I. Krummenacher, H. Braunschweig, U. Radius, Eur. J. Inorg. Chem. 2020, 281-291.
Abstract
The reaction of one and two equivalents of the N-heterocyclic carbene IMes (IMes = 1,3-bis(2,4,6-trimethyl-phenyl)imidazolin-2-ylidene) or the cyclic (alkyl)(amino)carbene cAACMe (cAACMe = 1-(2,6-diisopropyl-phenyl)-3,3,5,5-tetra-methylpyrrolidin-2-ylidene) with [TiCl4] in n-hexane results in the formation of mono- and bis-carbene complexes [TiCl4(IMes)] 1, [TiCl4(IMes)2] 2, [TiCl4(cAACMe)] 3, and [TiCl4(cAACMe)2] 4, respectively. For comparison, the titanium(IV) NHC complex [TiCl4(IiPrMe)] 5 (IiPrMe = 1,3-diisopropyl-4,5-dimethyl-imidazolin-2-ylidene) has been synthesized and structurally characterized. The reaction of [TiCl4(IMes)] 1 with PMe3 affords the mixed substituted complex [TiCl4(IMes)(PMe3)] 6. The reactions of [TiCl3(THF)3] with two equivalents of the carbenes IMes and cAACMe in n-hexane lead to the clean formation of the titanium(III) complexes [TiCl3(IMes)2] 7 and [TiCl3(cAACMe)2] 8. Compounds 1-8 have been completely characterized by elemental analysis, IR- and multinuclear NMR spectroscopy and for 2-5, 7 and 8 by X-ray diffraction. Magnetometry in solution, EPR and UV-Vis spectroscopy and DFT calculations performed on 7 and 8 are indicative of a predominantly metal-centered d1-radical in both cases.
U. S. D. Paul, M. J. Krahfuß, U. Radius, Chem. Unserer Zeit 2019, 53, 212-223.
Abstract
The very important ligand class of carbenes has been expanded by very versatile new representatives, the cyclic (alkyl)(amino)carbenes. Due to their steric and electronic properties, cAACs are suitable ligands in transition metal chemistry, e.g. for the synthesis of catalysts and the stabilization of metals in unusual oxidative states. In main group chemistry, they are used as reactive organocatalysts to activate small molecules or enthalpic strong bonds. In this review, some examples of ongoing research in main group as well as transition metal chemistry are discussed and meanwhile some basic concepts of organic and inorganic chemistry are presented (instructive review is written in German!).
Seit der ersten Synthese von cyclischen (Alkyl)(amino)carbenen (cAACs) im Jahre 2005 befindet sich diese Klasse von Carbenen auf einem Siegeszug durch die Hauptgruppenelement- und Übergangsmetall-Chemie. CAACs gehören zu den stärksten nukleophilen (σ-Donor) und gleichzeitig zu den stärksten elektrophilen (π-Akzeptor) Singulett-Carbenen, die bislang bekannt sind. Diese Eigenschaften führen zu einer Vielzahl von Anwendung dieser Moleküle, wie der Stabilisierung hochreaktiver Katalysatoren, der Stabilisierung reaktiver Verbindungen oder der Aktivierung kleiner Moleküle. In dieser Übersicht für Schüler und Studenten werden cAACs vorgestellt und einige dieser neuen Entwicklungen erörtert.
J. Lorkowski, M. Krahfuß, M. Kubicki, U. Radius, C. Pietraszuk, Chem. Eur. J. 2019, 25, 11365-11374.
Abstract
Tricky cyclic (Amino)(Aryl)Carbenes (cAArCs): Cyclic (amino)(aryl)carbenes (cAArCs) based on the isoindoline core were successfully generated in situ via a-elimination of 3-alkoxyisoindolines at high temperatures or by deprotonation of isoindol-2-ium chlorides with sodium or copper(I) actetates at low temperatures. 3-Alkoxy-isoindolines 2a,b-OR (R = Me, Et, iPr) have been prepared in high yields by treatment of solutions of 2-aryl-1,1-diphenylisoindol-2-ium triflates 1a,b-OTf (a: aryl = 2,6-diisopropylphenyl- [Dipp]; b: 2,4,6-trimethylphenyl [Mesityl-, Mes]) in the corresponding alcohol ROH with NEt3 at room temperature, and the reaction of 2a,b-OMe in diethyl ether with a tenfold excess of hydrochloric acid led to the isolation of the isoindol-2-ium chlorides 1a,b-Cl in high yields. Thermally generated cAArC reacts with sulfur to form the thioamide 3a. Without any additional trapping reagent in situ generation of 1,1-diphenylisoidolin-3-ylidenes does not lead to the isolation of these compounds, but to reaction products of the insertion of the carbene carbon atom into an ortho C-H bond of a phenyl substituent, followed by ring expansion reaction to give the anthracene derivatives 9-N(H)aryl-10-Ph-C14H8 4a,b (a: Dipp; b: Mes). These compounds are conveniently synthesized by deprotonation of the isoindol-2-ium chlorides with sodium acetate in high yields. Deprotonation of 1a-Cl with copper(I) acetate at low temperatures afforded a mixture of 4a and the corresponding cAArC copper(I) chloride 5a, and allowed the isolation and structural characterization of a first example of a cAArC copper complex of general formula [(cAArC)CuCl].
L. Kuehn, D. G. Jammal, K. Lubitz, T. B. Marder, U. Radius, Chem. Eur. J. 2019, 25 ,9514-9521; „Hot Paper“
Abstract
NHC-nickel complexes are efficient catalysts for the C‑Cl bond borylation of aryl chlorides using NaOAc as a base and B2pin2 as the boron source. The catalysts [Ni2(ICy)4(μ-(η2:η2)-COD)] 1 (ICy = 1,3-dicyclohexylimidazolin-2-ylidene; COD = cyclooctadiene;), [Ni(ICy)2(η2-C2H4)] 2 and [Ni(ICy)2(η2-COE)] 3 (COE = cyclooctene) compare well with other nickel catalysts reported previously for aryl chloride borylation with the advantage that no further ligands have to be added to the reaction. Borylation also proceeds with B2neop2 as the boron source. Stoichiometric oxidative addition of different aryl chlorides to complex 1 is highly selective affording trans-[Ni(ICy)2(Cl)(Ar)] (Ar = 4-(F3C)C6H4, 11; 4-(MeO)C6H4, 12; C6H5, 13; 3,5-F2C6H3, 14).
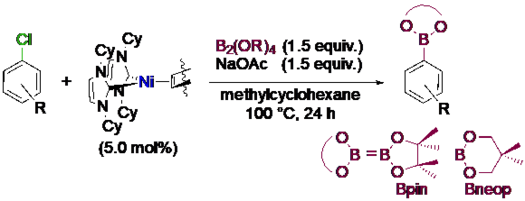
J. H. J. Berthel, L. Tendera, M. W. Kuntze-Fechner, L. Kuehn, U. Radius, Eur. J. Inorg. Chem. 2019, 3061-3072.
Abstract
We reported recently on the reactivity of the dinuclear complex [{Ni(iPr2Im)2}2(μ-(η2:η2)-COD)] (iPr2Im = 1,3‑di‑isopropyl-imidazolin-2-ylidene, COD = 1,4-cyclooctadiene) in Hiyama- and Negishi-type cross-coupling reactions as well as on the synthesis and stability of several novel nickel fluoroaryl alkyl complexes (Eur. J. Inorg. Chem. 2019, 1767-1775). Herein we expanded this study to the synthesis of different NHC-stabilized nickel olefin, alkyne, alkyl and cyanido complexes starting from trans-[Ni(iPr2Im)2Br2], trans-[Ni(iPr2ImMe)2Br2], trans-[Ni(Me2Im)2I2], and trans-[Ni(Me2ImMe)2Br2] as synthetically useful nickel (II) precursors.
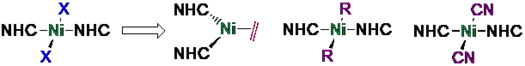
K. Lubitz, U. Radius, Organometallics 2019, 38, 2558-2572.
Abstract
We present herein studies on the reactivity of complexes of the type [(η5-C5H5)Co(NHC)(olefin)] with internal and terminal alkynes. Surprisingly, the half sandwich complexes [(η5-C5H5)Co(iPr2Im)(η2-C2H4)] (1, iPr2Im = 1,3-diisopropyl-imidazolin-2-ylidene) and [(η5-C5H5)Co(Dipp2Im)(η2-C2H3SiMe3)] (2, Dipp2Im = 1,3-bis(2,6-diisopropyl-phenyl)imidazolin-2-ylidene), are not good catalysts for the [2+2+2] cyclotrimerization of alkynes (Vollhardt cyclization). Stoichiometric reactions of 1 and 2 with diphenylacetylene, phenylacetylene and p-tolylacetylene have shown that the behavior of internal and terminal alkynes towards complexes [(η5-C5H5)Co(NHC)(olefin)] is different. The reaction of 1 and 2 with diphenylacetylene leads to the formation of [(η5-C5H5)Co(R12Im)(η2-C2Ph2)] (R1= iPr 3, Dipp 4). Complex 4 reacts at elevated temperatures with another equivalent of diphenylacetylene to afford [(η5-C5H5)Co(η4-C4Ph4)] (5), which seems to be the dead end of the catalysis. Complexes 1 and 2 react with phenylacetylene and p-tolylacetylene with coupling of the NHC and the alkyne in the coordination sphere of the cobalt atom. Depending on the NHC ligand used, the complexes [(η5-C5H5)Co(–C{R}=C{H}{Dipp2Im})] (R = Ph 6; Tol 7) and [(η5-C5H5)Co(η4-{(RER-NHC)C4H2R2}] (R = Ph (8), Tol (9)) are formed. In course of these reactions the NHCs migrate from the metal atom to the alkyne α carbon atom. For the NHC iPr2Im a second equivalent of the alkyne reacts with 1 with ring opening of the NHC (see Scheme).
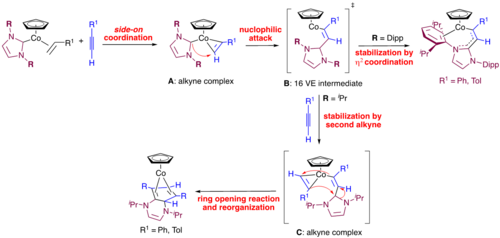
J. H. J. Berthel, M. W. Kuntze-Fechner, U. Radius, Eur. J. Inorg. Chem. 2019, 2618-2623.
Abstract
The reactivity of nickel tetracarbonyl [Ni(CO)4] with the N-Heterocyclic Carbenes (NHCs) 1,3-di-iso-propylimidazolin-2-ylidene (iPr2Im) and 1,3-di(isopropyl)-4,5-di(methyl)imidazolin-2-ylidene (iPr2ImMe) is reported. The reaction of [Ni(CO)4] with two equiv. iPr2Im and iPr2ImMe and one equiv. iPr2ImMe leads to the formation of the complexes [Ni(iPr2Im)2(CO)2] 2, [Ni(iPr2ImMe)2(CO)2] 3, and [Ni(iPr2ImMe)(CO)3] 4. The reaction of [Ni(CO)4] with one equiv. (or less) iPr2Im affords the triangular 44 VE (valence electron) cluster complex [Ni3(iPr2Im)3(µ2-CO)3(µ3-CO)] 1, a NHC-stabilized analogue of the parent neutral nickel Chini cluster [Ni3(CO)3(µ2-CO)3].
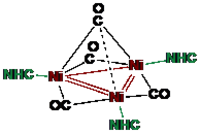
M. W. Kuntze-Fechner, C. Kerpen, D. Schmidt, M. Häring, U. Radius, Eur. J. Inorg. Chem. 2019, 1767-1775.
Abstract
The reactivity of [Ni(iPr2Im)4(µ-COD)] 1 (iPr2Im = 1,3‑di‑isopropyl-imidazolin-2-ylidene, COD = 1,4-cyclooctadiene) in Hiyama- and Negishi-type cross-coupling reactions of perfluoroaromatics as well as the synthesis and investigations concerning the stability in solution of several novel nickel fluoroaryl alkyl complexes is reported.
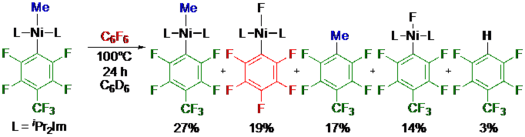
Y.-M. Tian, X.-N. Guo, M. Kuntze-Fechner, I. Krummenacher, H. Braunschweig, U. Radius, A. Steffen, T. B. Marder, J. Am. Chem. Soc. 2018, 140, 17612-17623.
Abstract
We present herein a highly selective and general photocatalytic C-F borylation protocol that employs a rhodium biphenyl complex as a triplet sensitizer and the nickel catalyst [Ni(IMes)2] (IMes = 1,3-dimesitylimidazolin-2-ylidene) for the C-F bond activation and defluoroborylation. This tandem catalyst system operates with visible (blue, 400 nm) light and achieves borylation of a wide range of fluoroarenes with B2pin2 at room temperature in excellent yields and with high selectivity. Mechanistic studies suggest that the exceptionally long-lived triplet excited state of the Rh biphenyl complex used as the photosensitizer allows for efficient triplet energy transfer to trans-[NiF(ArF)(IMes)2], which leads to dissociation of one of the NHC ligands. This contrasts with the majority of current photocatalytic transformations, which employ transition metals as excited state single electron transfer agents. We have previously reported that C(arene)-F bond activation with [Ni(IMes)2] is facile at room temperature, but that the transmetalation step with B2pin2 is associated with a high energy barrier. Thus, this triplet energy transfer ultimately leads to a greatly enhanced rate constant for the transmetalation step and thus for the whole borylation process.
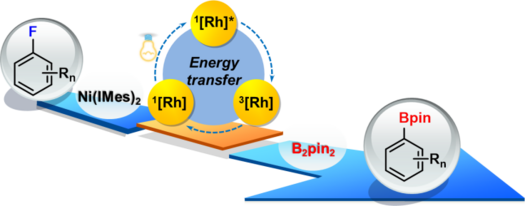
H. Schneider, A. Hock, A. D. Jaeger, D. Lentz, U. Radius, Eur. J. Inorg. Chem. 2018, 4031-4043.
Abstract
We present herein the utilization of NHC-stabilized alane adducts of the type (NHC)∙AlH3 (NHC = Me2Im 1, Me2ImMe 2, iPr2Im 3, iPr2ImMe 4, Dipp2Im 5) and (NHC)∙AliBu2H (NHC = iPr2Im 6, Dipp2Im 7) as novel hydride transfer reagents in the hydrodefluorination (HDF) of different fluoroaromatics and hexafluoropropene. In dependence on the alane adduct used, HDF of pentafluoropyridine to 2,3,5,6-tetrafluoropyridine from 15-99 % is observed.
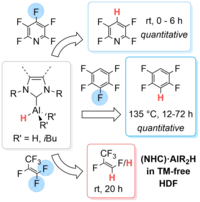
K. Lubitz, V. Sharma, S. Shukla, J. H. J. Berthel, H. Schneider, C. Hoßbach, U. Radius, Organometallics 2018, 37, 1181 - 1191.
Abstract
The synthesis of novel asymmetrically substituted cobalt complexes of the type [Co(CO)(NO)(NHC)(PR3)] (NHC = iPr2Im; PR3 = PMe3 (1), PEt3(2), PHiPr2 (3); PR3 = PMe3; NHC = Me2ImMe (4), MeiPrIm (5), MetBuIm (6), iPr2ImMe (7); R2Im: 1,3-dialkylimidazolin-2-ylidene) is reported. These complexes are stabilized by a N-heterocyclic carbene (NHC), a phosphine, a carbonyl and a nitrosyl ligand and have been synthesized from the reaction of a NHC-substituted precursor of the type [Co(CO)2(NO)(NHC)] and the corresponding phosphine. The synthesis of [Co(CO)(NO)(MetBuIm)(PMe3)] (6) and [Co(CO)(NO)(iPr2ImMe)(PMe3)] (7) proceeds in a thermal reaction already at room temperature by quantitative replacement of one carbonyl with a phosphine ligand. All the other complexes were synthesized using photochemical conditions. The complexes 1 – 6 have been characterized by elemental analysis, IR spectroscopy, multinuclear NMR spectroscopy, and in some cases by X-ray crystallography. All complexes are volatile, stable upon sublimation and decompose readily in a stepwise manner at elevated temperature. The complex [Co(CO)(NO)(iPr2Im)(PMe3)] (1) as well as cobalt complexes that were reported earlier i.e. [Co(CO)(NO)(iPr2Im)2], [Co(CO)(NO)(MetBuIm)2] and [Co(CO)2(NO)(iPr2Im)] are evaluated and have been successfully applied in the deposition of cobalt-based thin films.
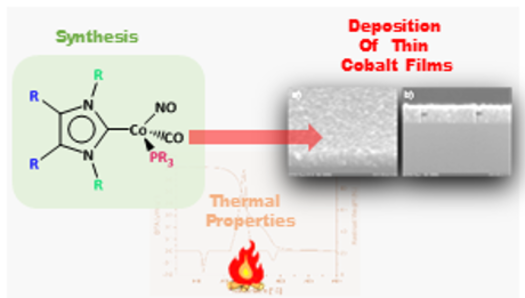
A. F. Eichhorn, L. Kuehn, T. B. Marder, U. Radius, Chem. Commun. 2017, 53, 11694-11696.
Abstract
We report herein the room temperature insertion of the carbene carbon atom of the cyclic (alkyl)(amino) carbene cAACMe into the B-B single bonds of the diboron(4) compounds B2pin2, B2cat2, B2neop2, and B2eg2 (pin = pinacolato, cat = catecholato, eg = ethyleneglycolato, neop = neopentylglycolato).

H. Schneider, A. Hock, R. Bertermann, U. Radius, Chem Eur. J. 2017, 23, 12387-12398.
Dedicated to Dieter Fenske
Abstract
The synthesis of the mono NHC alane adducts of the type (NHC)·AlH3 (NHC = Me2Im 1, Me2ImMe 2, iPr2Im 3 and 3-d3, iPr2ImMe 4, Dipp2Im 10) and (NHC)·AliBu2H (NHC = iPr2Im 11, Dipp2Im 12) as well as their reactivity towards different types of carbenes is presented. While the mono NHC adducts remain stable at elevated temperatures, ring expansion reaction occurs when (iPr2Im)·AlH3 3 is reacted with a second equivalent of the carbene iPr2Im to give (iPr2Im)·AlH(RER-iPr2ImH2) 6. In 6, {(iPr2Im}AlH} is inserted into the NHC ring. In contrast, ring opening is observed with sterically more demanding Dipp2Im with formation of (iPr2Im)·AlH2(ROR-Dipp2ImH2)H2Al·(iPr2Im) 9. In 9, two {(iPr2Im)·AlH2} moieties stabilize the ring opened Dipp2Im. If two hydridic sites are blocked, the adducts are stable with respect to further ring expansion or ring opening, as exemplified by the adducts (iPr2Im)·AliBu2H 11 and (Dipp2Im)·AliBu2H 12. The reaction of (NHC)·AlH3 and (iPr2Im)·AliBu2H with cAACMe yields instead of ligand substitution, ring expansion or ring opening the products of an insertion of the carbene carbon atom into the Al–H bond (NHC)·AlH2/iBu2(cAACMeH) 13–18.
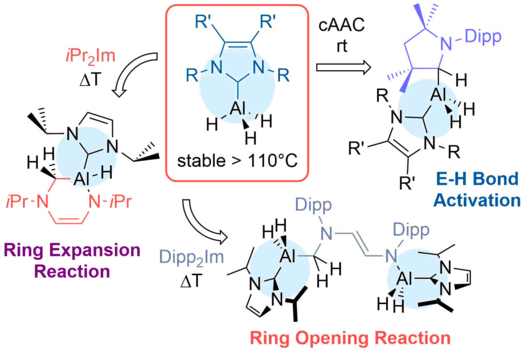
U. S. D. Paul, U. Radius, Eur. J. Inorg. Chem. 2017, 3362-3375.
Abstract
The first synthesis of cyclic (alkyl)(amino) carbenes (cAACs) was reported in 2005, and since then this class of carbenes is on a victory tour in main group element and transition metal chemistry. CAACs are easy to synthesize and among the most nucleophilic (σ-donating) and simultaneously most electrophilic (π-accepting) carbenes known to date. These properties led to a vast number of applications of cAACs in main group element chemistry, for example in the activation of small molecules and enthalpically strong bonds, as well as in the stabilization of highly reactive main group element compounds. They also proved to possess outstanding ligand properties in transition metal chemistry, for example in the stabilization of unusual low-valent transition metal complexes leading to enormous highly active but robust catalysts. Herein, we give a brief overview of cAAC-ligated transition metal compounds in mainly low oxidation states with an emphasis on nickel complexes and an account on selected recent developments of the use of cAACs in coordination chemistry and catalysis.
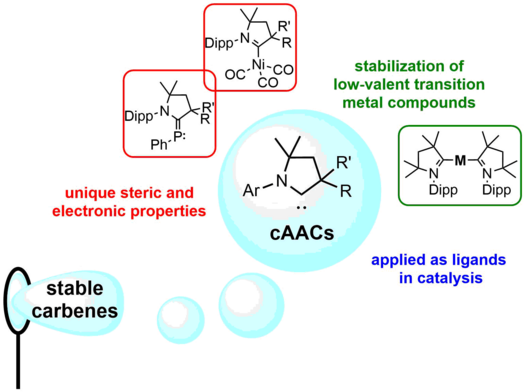
A. F. Eichhorn, S. Fuchs, M. Flock, T. B. Marder, U. Radius, Angewandte Chemie 2017, 129, 10343-10347; Angewandte Chemie Int. Ed. 2017, 56, 10209-10213.
Dedicated to Roald Hoffmann
Abstract
Carbon is the better metal! Just as their isolobal complex fragments C2v-d8-ML4 or C2v-d10-ML2, the carbene cAACMe reacts with selected aryl boronate esters with reversible oxidative addition at room temperature. This reversible, metal-free B-C bond activation at carbon might lead to the development of novel organocatalytic reactions with carbon atom catalysts.

Synthesis and Reactivity of Cyclic (Alkyl)(Amino)Carbene stabilized Nickel Carbonyl Complexes
U. S. D. Paul, U. Radius, Organometallics 2017, 36, 1398-1407.
Abstract
Cyclic (alkyl)(amino)carbenes cAACcy (1b) and cAACmenthyl (1c) react with [Ni(CO)4] to give the 18 VE complexes [Ni(CO)3(cAACcy)] (2b) and [Ni(CO)3(cAACmenthyl)] (2c). With these in hands, the donor-strength and the steric profile of the respective cAAC ligands were evaluated. CAACcy and cAACmenthyl possess similar overall-donating properties (TEP = 2046 (1b) and 2042 (1c)) as common NHCs, though they are also known to be better π-acceptors. 3,3-diamino-2-aryloxyacrylimidamide 3b, arising from the reaction of cAACcy (1b) with released CO molecules, was obtained as side-product of CO substitution reactions at nickel carbonyls. In contrast to cAACmenthyl (%Vbur = 42), the sterically less encumbered cAACcy (%Vbur = 38) undergoes a subsequent CO substitution at [Ni(CO)3(cAACCy)] (2b) to afford the 16 VE complex [Ni(CO)(cAACcy)2] (4b). Treatment of both [Ni(CO)3(cAACmethyl)] (2a) and [Ni(CO)(cAACmethyl)2] (4a) with allyl bromides led to the formation of cAAC-stabilized allyl nickel complexes [NiBr(η3-H2C=CH-CH2)(cAACmethyl)] (5a) and [NiBr(η3-H2C=CH-CMe2)(cAACmethyl)] (5b). The chloro complex [NiCl(η3-H2C=CMe-CH2)(cAACmethyl)] (6) was synthesized from [Ni(COD)2] (COD = 1,5-cyclooctadiene) by consecutive treatment with allyl chloride and cAAC. The allyl halide complexes 5-6 are thermally labile and decompose in solution already within a few hours at room temperature. One of the decomposition products, the dinuclear nickel complex [Ni2(μ-Br)2(η3-(cAACmethyl)=CH–CH(CH3))2] (7), was crystallographically characterized.
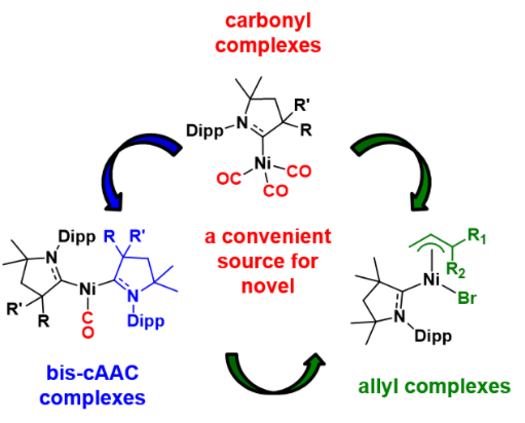
H. Schneider, D. Schmidt, A. Eichhöfer, M. Radius, F. Weigend, U. Radius, Eur. J. Inorg. Chem. 2017, 2600-2616.
Abstract
We present the synthesis of a series of NHC stabilized iron(II) complexes of the type [Fe(NHC)2Mes2] and [Fe(NHC)Mes2], starting from [Fe2Mes4] and the NHC. In dependence on the steric demand of the NHC used either tetrahedral [Fe(Me2Im)2Mes2] 1 and [Fe(MeiPrIm)2Mes2] 2, square planar trans-[Fe(iPr2Im)2Mes2] 3, or trigonal planar [Fe(R2Im)Mes2] (R = tBu 6, Dipp 7, Dipp2H2 8) were isolated. Three coordinated complexes [Fe(R2Im)Mes2] (R = Me 4, iPr 5) of smaller carbenes were synthesized via ligand exchange of [Fe(NHC)2Mes2] and [Fe2Mes4]. The reactivity of trans-[Fe(iPr2Im)2Mes2] 3 towards small molecules and element hydrogen compounds was investigated. The reaction of 3 and 2 with dioxygen led to the tetrameric cluster complex [{Fe(R2Im)(OMes)(µ2-O)}4] 9 and 10. The reaction of 3 with CO affords [Fe(iPr2Im)2(CO)3] 11. Reactions of 3 with E–H compounds produced with substitution of the mesityl ligands the complexes [Fe(iPr2Im)2(OtBu)2] 12, [Fe(iPr2Im)2(OPh)2] 13, [Fe(iPr2Im)2(SPh)2] 14, [Fe(iPr2Im)2(NHPh)2] 15, cis-[Fe(iPr2Im)2{o-C6H4(NH)2}] 16, [Fe(iPr2Im)2(PHPh)2] 17, [Fe(iPr2Im)2(PPh2)2] 18, trans-[Fe(Me2Im)2(SnnBu3)2] 19 and trans-[Fe(iPr2Im)2(SnnBu3)2] 20. The complexes 16, 17, 19, and 20 adopt square planar structures. The reaction of 3 with phenyl silane afforded crystallographically characterized [Fe(iPr2Im)2(µ-H)3(SiPh2H){SiH2(iPr2Im)}] 21.
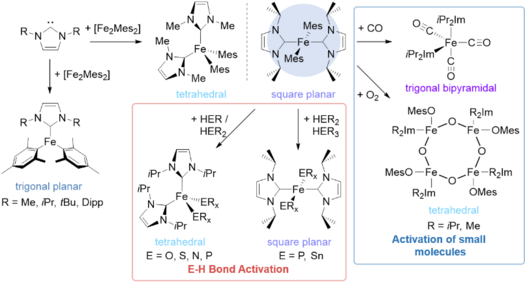
M. Eck, S. Würtemberger-Pietsch, A. Eichhorn, J. H. J. Berthel, R. Bertermann, U. S. D. Paul, H. Schneider, A. Friedrich, C. Kleeberg, U. Radius, T. B. Marder, Dalton Transactions 2017, 46, 3661-3680.
Abstract
In this detailed study we report on the structures of the widely employed diboron(4) compounds bis(pinacolato)diboron (B2pin2) and bis(neopentyl glycolato)diboron (B2neop2), as well as bis(ethylene glycolato)diboron (B2eg2) and tetrakis(dimethylamino)diboron (B2(NMe2)4), and their reactivity, along with that of bis(catecholato)diboron (B2cat2) with backbone saturated and backbone unsaturared N-heterocyclic carbenes (NHCs) of different steric demand. Depending on the nature of the diboron(4) compound and the NHC used, Lewis-acid/Lewis-base adducts or NHC ring expansion products stemming from B–B and C–N bond activation have been observed. The corresponding NHC adducts and NHC ring-expanded products were isolated and characterised, via solid-state and solution NMR spectroscopy and X-ray diffraction. In general, we observed B–B bond and C–N bond activation at low temperature for B2eg2, at room temperature for B2neop2 and at higher temperature for B2cat2. The reactivity strongly depends on steric effects of the NHCs and the diboron(4) compounds, as well as on the corresponding Lewis-basicity and Lewis-acidity. Our results provide profound insight into the chemistry of these diboron(4) reagents with the nowadays ubiquitous NHCs, the stability of the corresponding NHC adducts and on B–B activation using Lewis-bases in general. We demonstrate that B–B bond activation may be triggered even at temperatures as low as -40 °C to -30 °C without any metal species involved. For example, the reactions of B2eg2 with sterically less demanding NHCs such as Me2ImMe and iPr2Im in solution led to the corresponding ring-expanded products at low temperatures. Furthermore, boronium [L2B(OR)2]+ and borenium [LB(OR)2]+ cations have been observed from the reaction of the bis-borate B2eg3 with the NHCs iPr2Im and Me2ImMe, which led to the conclusion that the activation of bis-borates with NHCs (or Lewis-bases in general) might be a facile and simple route to access boron electrophiles. Both boron-based nucleophiles and electrophiles are important for applications in organic synthesis.
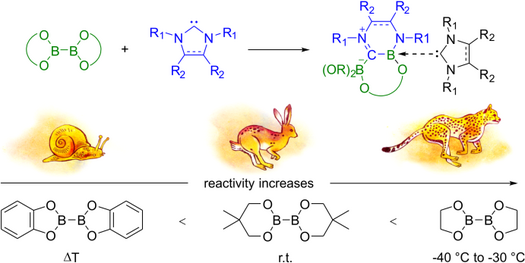
U. S. D. Paul, U. Radius, Chem Eur. J 2017, 23, 3993-4009.
Abstract
C-F and C-H bond activation reactions of polyfluoroaromatics at the cyclic (alkyl)(amino)carbene (cAAC) cAACmethyl (1) is reported. Studies on the C–F bond activation using the cAAC-stabilized nickel(0) complex [Ni(cAACmethyl)2] (2) have shown that 2 does not react with fluorinated arenes. However, these investigations led to the observation of C–F bond cleavage of perfluorinatied arenes by the carbene ligand cAACmethyl (1) itself. The reaction of with C6F6, C6F5–C6F5, C6F5–CF3, and C5F5N afforded the insertion products of cAAC into one of the C-F bonds of the substrate, i.e. the C-F bond activation products (cAACmethyl)F(Arf) (Arf = C6F5 4a, C6F4–C6F5 4b, C6F4–CF3 4c, C5F5N 4d). These products decompose readily upon heating to 80 °C within a few hours in solution with formation of ionic iminium salts [(cAACmethyl)(Arf)][X] 6a-d or neutral alkenyl perfluoroaryl imine complexes 7a-d. The compounds (cAACmethyl)F(Arf) 4a-d readily transfer fluoride, which has been exemplified by the fluoride transfer of all compounds using BF3 etherate as fluoride acceptor. Fluoride transfer has also been achieved starting from (cAACmethyl)F(C6F4–CF3) (4c) or (cAACmethyl)F(C5F4N) (4d) to other selected substrates such as trimethyl chloro silane, benzoyl chloride and tosyl chloride. Instead of C-F bond activation, insertion of the cAAC into the C-H bond was observed if 1 was treated with the partially fluorinated arenes C6F5H, 1,2,4,5-C6F4H2, 1,3,5-C6F3H3, and 1,3-C6F2H4. The compounds (cAACmethyl)H(Arf) (Arf = C6F5 12e, 2,3,4,5-C6F4H 12f, 2,4,6-C6F3H2 12g and 2,6-C6F2H3 12h) have been isolated in good yields and have been characterized including X-ray analysis. Fluorobenzene C6FH5 ((pKa ≈ 37), the least C–H acidic fluoroarene used in this study, does not react. In order to investigate the scope and limitations of this type of cAAC C-H bond activation reaction, cAACmethyl (1) was treated with several other reagents of different C–H acidity such as imidazolium salts, imidazoles, esters and trimethylphosphine. These investigations led to the isolation and characterization of the compounds [(cAACmethyl)H(R2ImMe2)]X (13a,b), (cAACmethyl)H(ImR2) (14a-c), (cAACmethyl)H(CH(COOCH3)2) (15b) and (cAACmethyl)H(CH2–PMe2) (16). Deprotonation of [(cAACmethyl)H(Me2ImMe2)][BF4] (13a) at the cAAC carbon atom using KHMDS as a base led to isolation and structural characterization of the cAACmethyl-NHC heterodimer (17).
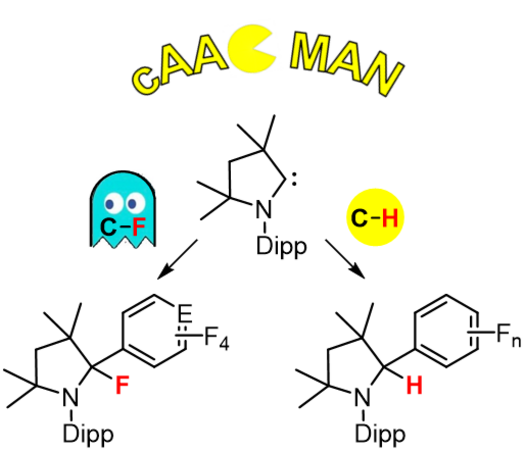
S. Würtemberger-Pietsch, H. Schneider, T. B. Marder, Udo Radius, Chem Eur. J. 2016, 22, 13032-13036.
Abstract
It’s all about the Carbene! The isolation and detailed structural characterization of a series of adducts of catecholborane (HBcat) with backbone unsaturated N-heterocyclic carbenes (NHCs), a B-H activation product with a cyclic (alkyl)(amino)carbene CAACMe, and a ring expanded product with a backbone saturated NHC is reported. The mono-NHC adducts and the B-H activation product are stable at higher temperatures, whereas the product of the reaction of HBcat with the saturated NHC undergoes a rearrangement even at room temperature, forming a six-membered heterocyclic ring.
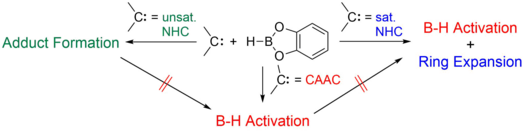
F. Hering, J. H. J. Berthel, K. Lubitz, U. S. D. Paul, H. Schneider, M. Härterich, U. Radius, Organometallics 2016, 35, 2806-2821.
Abstract
The synthesis of a series of cobalt NHC complexes of the types [Co(NHC)2(CO)(NO)] (NHC = iPr2Im (2), nPr2Im (3), Cy2Im (4), Me2Im (5), iPr2ImMe (6), Me2ImMe (7), MeiPrIm (8), MetBuIm (9); R2Im = 1,3-dialkylimidazolin-2-ylidene) and [Co(NHC)(CO)2(NO)] (NHC = iPr2Im (13), nPr2Im (14), Me2Im (15), iPr2ImMe (16), Me2ImMe (17), MeiPrIm (18), MetBuIm (19)) from the reaction of the NHC with [Co(CO)3(NO)] (1) is reported. These complexes have been characterized using elemental analysis, IR spectroscopy, multinuclear NMR spectroscopy, and in many cases by X-ray crystallography. Bulky NHCs tend to form the mono-NHC-substituted complexes [Co(NHC)(CO)2(NO)], even from the reaction with an stoichiometric excess of the NHC, as demonstrated by the synthesis of [Co(Dipp2Im)(CO)2(NO)] (11), [Co(Mes2Im)(CO)2(NO)] (12), and [Co(MecAAC)(CO)2(NO)] (20). For tBu2Im a preferred coordination via the NHC backbone (“abnormal” coordination at the 4-position) was observed and the complex [Co(tBu2aIm)(CO)2(NO)] (10) was isolated. All of these complexes are volatile, are stable upon sublimation and prolonged storage in the gas phase, and readily decompose at higher temperatures. Furthermore, DTA/TG analyses revealed that the complexes [Co(NHC)2(CO)(NO)] are seemingly more stable toward thermal decomposition in comparison to the complexes [Co(NHC)(CO)2(NO)]. We thus conclude that the cobalt complexes of the type [Co(NHC)(CO)2(NO)] and [Co(NHC)2(CO)(NO)] have potential for application as precursors in the vapor deposition of thin cobalt films.
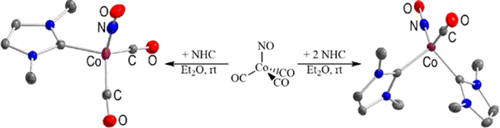
NHC Nickel-Catalyzed Suzuki-Miyaura Cross-Coupling Reactions of Arylboronate Esters with Perfluorobenzenes
J. Zhou, J. H. J. Berthel, A. Friedrich, T. B. Marder, U. Radius, J. Org. Chem. 2016, 81, 5789-5794.
Abstract
An efficient Suzuki-Miyaura cross-coupling reaction of perfluorinated arenes with aryl boronate esters using NHC nickel complexes as catalysts is described. The efficiencies of different boronate esters (p-tolyl-Beg, p-tolyl-Bneop, p-tolyl-Bpin, p-tolyl-Bcat) and the corresponding boronic acid (p-tolyl-B(OH)2) in this type of cross-coupling reaction was evaluated (eg = ethyleneglycolato; neop = neopentylglycolato; pin = pinacolato, cat = catecholato). Aryl-Beg was shown to be the most reactive boronate ester among those studied. The use of CsF as an additive is essential for an efficient reaction of hexafluorobenzene with aryl neopentylglycolboronates.
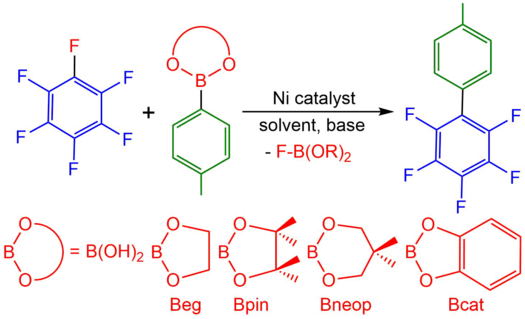
Cyclic (Alkyl)(Amino)Carbene Complexes of Rhodium and Nickel and their Steric and Electronic Parameters
U. S. D. Paul, C. Sieck, M. Hähnel, K. Hammond, T. B. Marder, U. Radius, Chem Eur. J. 2016, 21, 11005 – 11014.
Abstract
N-Heterocyclic carbenes (NHCs) and cyclic (alkyl)(amino)carbenes (CAACs) are of great interest as their electronic and steric properties provide a unique class of ligands and organo-catalysts. Herein we report our studies on substitution reactions involving novel carbonyl complexes of rhodium and nickel to provide a deeper understanding of the fundamental electronic factors characterizing CAACmethyl (1), and compare it with the large array of data available for NHC and sterically more demanding CAAC ligands.
Iridium-Catalysed Dehydrocoupling of Aryl Phosphine-Borane Adducts: Synthesis and Characterisation of High Molecular Weight Poly(phosphinoboranes)
U. S. D. Paul, H. Braunschweig, U. Radius, Chem. Commun. 2016, 52, 8573-8576.
Abstract
The thermal dehydrogenative coupling of aryl phosphine-borane adducts with iridium complexes bearing a bis(phosphinite) pincer ligand is reported. This catalysis produces high molecular weight poly(phosphinoboranes) [ArPH–BH2]n (Ar = Ph, pTol, Mes). Furthermore, we investigated the reactivity of these pincer complexes towards primary phosphines and their respective borane adducts on a stoichiometric scale.
J. Zhou, M. Kuntze-Fechner, R. Bertermann, U. S. D. Paul, J. H. J. Berthel, A. Friedrich, Z. Du, T. B. Marder, U. Radius, J. Am. Chem. Soc. 2016, 138, 5250–5253.
Abstract
The [Ni(IMes)2]-catalyzed transformation of fluoroarenes into arylboronic acid pinacol esters via C−F bond activation and transmetalation with bis-(pinacolato)diboron (B2pin2) is reported. Various partially fluorinated arenes with different degrees of fluorination were converted into their corresponding boronate esters.
S. Würtemberger-Pietsch, U. Radius, T. B. Marder, Dalton Trans. 2016, 45, 5880–5895.
Abstract
N-Heterocyclic carbenes (NHCs) are widely used ligands and reagents in modern inorganic synthesis as well as in homogeneous catalysis and organocatalysis. However, NHCs are not always innocent bystanders. In the last few years, more and more examples were reported of reactions of NHCs with main-group elements which resulted in modification of the NHC. Many of these reactions lead to ring expansion and the formation of six-membered heterocyclic rings involving insertion of the heteroatom into the C–N bond and migration of hydrides, phenyl groups or boron-containing fragments. Furthermore, a few related NHC rearrangements were observed some decades ago. In this Perspective, we summarise the history of NHC ring expansion reactions from the 1960s till the present.
N. Arnold, S. Mozo, U. Paul, U. Radius, H. Braunschweig, Organometallics 2015, 34, 5709–5715.
Abstract
A series of iridium dihydroborate complexes [(tBuPOCOP)IrH(κ2-H2BHR)] (tBuPOCOP= κ3-C6H3-1,3-[OPtBu2]2; R = Mes = 2,4,6-Me3C6H2; R = Dur = 2,3,5,6-Me4C6H), [LIrH(κ2-H2BHDur)] (L = tBuPCP= κ3-C6H3-1,3-[CH2PtBu2]2, L = η5-C5Me5), and an osmium dihydroborate compound [OsH(κ2-H2BHDur)(CO)(PiPr3)2] have been prepared by using two different synthetic strategies. The first approach is based on direct borane coordination to the metal center whereas the second is based on a salt-elimination protocol using the lithium salts Li[H3BR] (R = Mes or Dur) and the corresponding metal halides. The compounds have been characterized by multinuclear NMR and IR spectroscopy and X-ray diffraction analysis. The results constitute the first syntheses of κ2-s:s-dihydroborate complexes featuring bulky aryl groups.
F. Hering, U. Radius, Organometallics 2015, 34, 3236–3245.
Abstract
The widely-held belief that N-heterocyclic carbenes (NHCs) act only as innocent spectator ligands is not always accurate, even in the context of well explored reactions. Ligand exchange in the conversion of [Pt(PPh3)2(η2-C2H4)] 3 to [Pt(iPr2Im)2] 2 depends critically on the particular reaction conditions employed, with slight changes leading to vastly different outcomes. In addition to [Pt(iPr2Im)2] 2, complexes [Pt(iPr2Im)(PPh3)(η2-C2H4)] 5 and trans-[Pt(iPr2Im)2(iPr-Im*)(H)] 6 were isolated and in the case of 6 fully characterized. Complex 5 represents the first mixed-olefin complex in transition metal chemistry containing both an NHC and a phosphine ligand. Chemical degradation of the NHC was shown to yield the new imidazole-2-yl iPr-Im* in 6. Therefore, the synthesis of [Pt(iPr2Im)2] 2 via metallic reduction of the ionic precursor [Pt(iPr2Im)3(Cl)]+Cl- 9 is favorable – a procedure adaptable to analogous palladium compounds. While [Pd(iPr2Im)3(Cl)]+Cl- 8 is the only product obtained from the reaction of iPr2Im and PdCl2, neutral [Pt(iPr2Im)2(Cl)2] 10, formed as a mixture of its two stereoisomers cis-10 and trans-10, is available through precise control of the stoichiometry in the reaction of PtCl2 and exactly two equivalents of iPr2Im.

H. Schneider, D. Schmidt, U. Radius, Chem. Commun. 2015, 51, 10138–10141.
Abstract
The dehydrogenative coupling of primary and secondary phosphines with the N-heterocyclic carbene iPr2Im (1,3-di-iso-propyl-imidazolin-2-ylidene) is reported. Dehydrogenation of R2PH affords diphosphines R2P–PR2. The reaction of iPr2Im with ArPH2 leads to NHC phosphinidene adducts iPr2Im=PAr and cyclic oligophosphines P4Ar4, P5Ar5 and P6Ar6, depending on the stoichiometry used. The NHC acts in these reactions as phosphine activator and hydrogen acceptor.
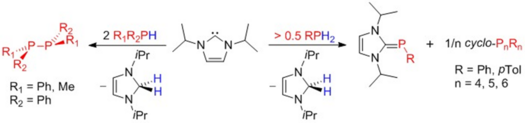
S. Pietsch, U. Paul, I. A. Cade, M. J. Ingleson, Udo Radius, T. B. Marder, Chem. Eur. J. 2015, 21, 9018–9021.
Abstract
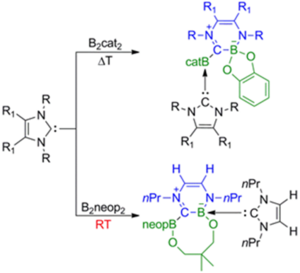
We report the isolation and detailed structural characterization, by solid-state and solution NMR spectroscopy, of the neutral mono- and bis-NHC adducts of bis(catecholato)diboron (B2cat2). The bis-NHC adduct undergoes thermally induced rearrangement, forming a 6-membered -B–C=N–C=C–N-heterocyclic ring via C–N bond cleavage and ring expansion of the NHC, whereas the mono-NHC adduct is stable. Bis(neopentylglycolato)diboron (B2neop2) is much more reactive than B2cat2 giving a ring expanded product at room temperature, demonstrating that ring expansion of NHCs can be a very facile process with significant implications for their use in catalysis.
H. Schneider, D. Schmidt, U. Radius, Chem. Eur. J. 2015, 21, 2793–2797.
Abstract
The reaction of iPr2Im (iPr2Im = 1,3-di-iso-propyl-imidazolin-2-ylidene) with diphenyldichlorosilane Ph2SiCl2 leads to the adduct (iPr2Im)-SiCl2Ph2 1. Prolonged heating of isolated 1 at 66 °C in THF affords the backbone tethered di(imidazolium) salt [(aHiPr2Im)2SiPh2]2+2Cl- 2 (“a” denotes “abnormal” coordination of the NHC), which can be synthesized in high yields in one step starting from two equivalents iPr2Im with Ph2SiCl2. Imidazolium salt 2 can be deprotonated in THF at room temperature using sodium hydride as a base and catalytic amounts of sodium tert-butoxide to give the stable N-heterocyclic dicarbene (aiPr2Im)2SiPh2 3, in which two NHCs are backbone tethered with a SiPh2 group. This easy-to-synthesize dicarbene 3 can be used as a novel ligand type in transition metal chemistry for the preparation of dinuclear NHC complexes, as exemplified by the synthesis of the homodinuclear copper(I) complex [{a(ClCu-iPr2Im)}2SiPh2] 4.
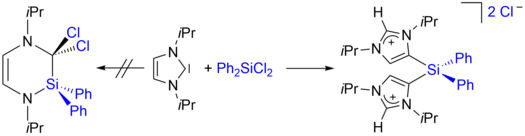
P. Hemberger, A. Bodi, J. H. J. Berthel, U. Radius, Chem. Eur. J. 2015, 21, 1434–1438.
Abstract
Ring expansion reactions (RER) of the N-heterocyclic carbene 1,3-dimethylimidazolin-2-ylidene was observed in the gas phase upon VUV photoexcitation. Similarly to RERs reported in the condensed phase for the reaction of NHCs with some main group element hydrides, depopulation of the NHC carbon atom seems to be the crucial initial step. In an ionization-mediated protonation, 1,3-dimethylimidazolin-2-ylidene forms an imidazolium ion, which is the rate limiting step on the pathway to two six-membered ring products, namely methylpyrimidinium and -pyrazinium ions. In order to unravel the reaction path, we have used imaging photoelectron photoion coincidence spectroscopy with vacuum ultraviolet (VUV) synchrotron radiation as well as high-level composite method calculations. Similarities and differences between the mechanism in the gas phase and in the condensed phase will be discussed.
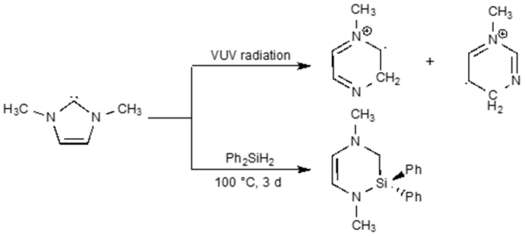
F. Hering, J. Nitsch, U. Paul, A. Steffen, F. M. Bickelhaupt, U. Radius, Chem. Sci. 2015, 6, 1426–1432.
Abstract
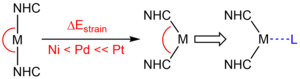
Synthesis, characterization and investigations on the reactivity of the novel metal basic, yet isolable 14 VE NHC-complexes [M0(iPr2Im)2] (M = Pd 1, Pt 2; iPr2Im = 1,3-di-isopropyl-imidazolin-2-ylidene; VE = valence electron; NHC = N-heterocyclic carbene) is reported and compared to the chemistry of the corresponding nickel complex. Quantum chemical analyses reveal that differences in the reactivity of group 10 NHC complexes are caused by differences in the rigidity and thus activation strain associated with bending the corresponding d10-[M(NHC)2] fragments during reaction. These results should have implications for the understanding of the fundamental steps in catalytic cycles, in which such complex fragments are employed.
D. Schmidt, T. Zell, T. Schaub, U. Radius, Dalton Trans. 2014, 43, 10816–10827.
Abstract
The unique reactivity of the nickel(0) complex [Ni2(iPr2Im)4(COD)] (1) (iPr2Im = 1,3-di-isopropyl-imidazolin-2-ylidene) towards hydrosilanes in stoichiometric and catalytic reactions is reported. A series of nickel hydrido silyl complexes cis-[Ni(iPr2Im)2(H)(SiHn-1R4-n)] (n = 1, 2) and nickel bis(silyl) complexes cis-[Ni(iPr2Im)2(SiHn-1R4-n)2] (n = 1, 2, 3) were synthesized by stoichiometric reactions of 1 with hydrosilanes HnSiR4-n, and fully characterized by X-ray diffraction and spectroscopic methods. These hydrido silyl complexes are examples, where the full oxidative addition step is hindered. They have, as a result of remaining Si–H interactions, remarkably short Si–H distances and feature a unique dynamic behavior in solution. Cis-[Ni(iPr2Im)2(H)(SiMePh2)] (cis-5) shows in solution at room temperature a dynamic site exchange of the NHC ligands, H-D exchange with C6D6 to give the deuteride complex cis-[Ni(iPr2Im)2(D)(SiMePh2)] (cis-5-D), and at elevated temperatures an irreversible isomerization to trans-[Ni(iPr2Im)2(D)(SiMePh2)] (trans-5-D). Reactions with sterically less demanding silanes give cis-configured bis(silyl) complexes under liberation of dihydrogen. These complexes display, similar to the hydrido silyl complexes, interestingly short Si–Si distances. Complex 1 reacts with 4 eq. HSi(EtO)3, in contrast to all the other silanes used in this study, to give the trans-configured bis(silyl) complex trans-[Ni(iPr2Im)2Ni(Si(OEt)3)2] (trans-12). The addition of two equivalents Ph2SiH2 to 1 results at elevated temperatures in the formation of the dinuclear complex [{(iPr2Im)Ni-m2-(HSiPh2)}2] (6). This diamagnetic, formal Ni(I) complex exhibits a long Ni–Ni bond in the solid state, as established by X-ray diffraction.

The capability of the electron rich {Ni(iPr2Im)2} complex fragment to activate Si–H bonds was applied catalytically in the deuteration of Et3Si–H to Et3Si–D employing C6D6 as convenient deuterium source. Furthermore, we show that 1 serves as a catalyst for the acceptorless dehydrogenative coupling of Ph2SiH2 to the corresponding disilane Ph2HSi–SiHPh2 and trisilane Ph2HSi–Si(Ph)2–SiHPh2, and the coupling of PhSiH3 to give a mixture of cyclic and linear polysilanes with high polydispersity (Mw = 1119; Mn = 924 ; Mw/Mn = 1.2). The capability of 1 to catalyze the formal reverse reaction as well is demonstrated by the hydrogenation of disilanes. The hydrogenation of the disilanes Ph2MeSi–SiMePh2 and PhMe2Si–SiMe2Ph to the corresponding hydrosilanes Ph2MeSi–H and PhMe2Si–H, respectively, proceeds effectively in the presence of 1 under very mild conditions (room temperature, 1.8 bar H2 pressure).
B. Zarzycki, F. M. Bickelhaupt, U. Radius, Dalton Trans. 2013, 42, 7468–7481.
Abstract
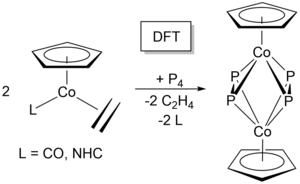
A full theoretical mechanistic investigation on the symmetrical degradation of P4 at the active complex fragments [(η5-C5H5)Co(L)] (L = CO, iPr2Im; iPr2Im = 1,3-di-iso-propylimidazolin-2-ylidene), which results in the formation of the complex [{(η5-C5H5)Co}2(µ, η2:2-P2)2] 9, is presented. The overall reaction mechanism is a complex, multistep process. Rate-determining steps of the reaction sequence are two consecutive dissociations of the co-ligands L, which induce the decisive structural rearrangements of the P4 unit. The choice of the co-ligand L (= CO, iPr2Im) influences the kinetic barrier as well as the energy balance of the overall reaction path significantly. The calculations further reveal a strong influence of the entropic effect on the overall reaction. As a consequence, the energy balance of the overall formation of 9 starting from [(η5-C5H5)Co(CO)] precursors is almost thermoneutral and has to overcome high kinetic barriers, whereas the reaction starting from [(η5-C5H5)Co(iPr2Im)] precursors with L = NHC is exothermic, features lower transition barriers with stabilized intermediates. From the direct comparison of both reaction coordinates it seems that the entropic effect of the co-ligands is even stronger than their electronic influence, as for both investigated systems the reactions' energy profiles are almost identical up to intermediate [{(η5-C5H5)Co(L)}2(μ,η2:2-P4)] 5 (L = CO, iPr2Im). After the formation of 5, the first CO dissociation step renders the reaction endothermic for L = CO, whereas in the case of NHC dissociation the reaction progresses exothermically. Energy decomposition analysis and fragment analysis provide a picture of the bonding mechanisms between the metal complex fragments and P4 in case of the most significant intermediates and the final product.
C. Hauf, J. E. Barquera-Lozada, P. Meixner, G. Eickerling, S. Altmannshofer, D. Stalke, T. Zell, D. Schmidt, U. Radius, W. Scherer, Z. Anorg. Allg. Chem. 2013, 639, 1996–2004.
Abstract
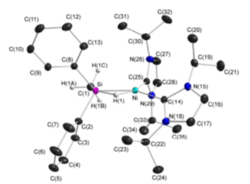
In general, it is assumed that the reaction between silanes and late transition metal fragments yields silyl hydride species as oxidative addition products. However, the silane complex Ni(iPr2Im)2(SiHMePh2) (iPr2Im = 1,3-diisopropylimidazolin-2-ylidene) (3a), might represent one of the rare systems where a stable η2-(Si–H)Ni intermediate of the oxidative addition process has been isolated. Indeed, 3a is characterized by an acute Si–Ni–H angle of 62.0(2)°, a rather short Si–H bond length of 1.992(6) Å and displaysa silicon-hydride cross peak in Si-H-HMQC 2D-NMR studies. We therefore selected the latter system for a combined experimental and theoretical charge density study to explore the electronic prerequisites which hinder the full completion of the oxidative addition step in transition metal silane complexes and cause the presence of remanent Si–H interactions in these species.
B. Zarzycki, T. Zell, D. Schmidt, U. Radius, Eur. J. Inorg. Chem. 2013, 2051–2058.
Abstract
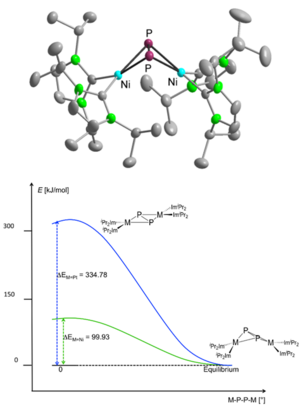
The reaction of [Ni2(iPr2Im)4(COD)] 1 with white phosphorus leads to dinuclear [{Ni(iPr2Im)2}2(µ,η2:2-P2)] 2 in excellent yield. This reaction represents the first example of a conversion of white phosphorus to a complex of the type [{L2Ni}2(µ,η2:2-P2)] and the first example for the formation of a complex of the type [{L2M}2(µ,η2:2-P2)] for a group 10 metal stabilized by two simple, non-chelating 2 electron donor ligands via the reaction of a suitable precursor with P4 in general. The X-ray molecular structure of 2 reveals a bent Ni2P2 core with a Ni-P-P-Ni dihedral angle in solid state of 102.95°. According to DFT calculations of the symmetrized model systems planar-D2h- and bent-C2v-[{Ni(iPr2Im)2}2(µ,η2:2-P2)], this deviation of the Ni2P2 core from planarity is caused by a second order Jahn Teller distortion. Calculations on the related platinum compound [{Pt(iPr2Im)2}2(µ,η2:2-P2)] confirm this type of bent structure for the higher congener with an even higher barrier to planarization as calculated for the nickel case. Energy decomposition analysis and fragment molecular orbital analysis further illustrate the bonding mechanisms in these complexes.
D. Schmidt, J. H. J. Berthel, S. Pietsch, U. Radius, Angew. Chem. 2012, 124, 9011–9015, Angew. Chem. Int. Ed. 2012, 51, 8881–8885. (Highlight: Nachr. Chem. 2012, 60, 1071)
Abstract
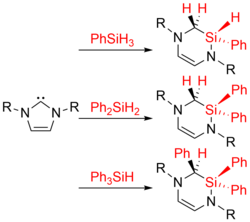
We report here the direct insertion of silylene moieties into the C-N bond of N-heterocyclic carbenes with ring expansion and formation of diaza-silinanes. These reactions are feasible using primary, secondary and tertiary silanes Ph4-nSiHn and a variety of NHCs, i.e. saturated and unsaturated NHCs with N-alkyl and N-aryl substituents. This ring expansion should be of general interest for the different areas of NHC-based chemistry of main group elements and transition metals, as well as for catalysis, especially if silanes are used in reactions with these species at higher temperature.
T. Zell, P. Fischer, D. Schmidt, U. Radius, Organometallics 2012, 31, 5065–5073.
Abstract
Complex [Ni2(iPr2Im)4(COD)] 1 (iPr2Im = 1,3-di-iso-propylimidazolin-2-ylidene) is a very efficient catalyst for the Suzuki-Miyaura cross coupling reaction of 4-bromotoluene with phenylboronic acid and also mediates the Ullmann-type homo cross coupling reaction of bromo benzene with a moderate efficiency. Stoichiometric reactions of complex 1 with aryl bromides (ArBr) at room temperature lead to mixtures of aryl bromo complexes of the type trans-[Ni(iPr2Im)2(Br)(Ar)] and the bis(bromo) complex trans-[Ni(iPr2Im)2(Br)2] 2. The complexes trans-[Ni(iPr2Im)2(Br)(Ar)] (for Ar = Ph 3, 4-MeC6H4 4, 4-Me(O)CC6H4 5, 4-MeOC6H4 6, 4-MeSC6H4 7, 4-Me2NC6H4 8, C5NH4 9) can be selectively synthesized by working at low temperatures and using high dilution of the starting materials. A major deactivation pathway for trans-[Ni(iPr2Im)2(Br)(Ar)] was identified in the presence of aryl bromides. This deactivation process includes (i) the formation of trans-[Ni(iPr2Im)2(Br)2] from trans-[Ni(iPr2Im)2(Br)(Ar)] and ArBr and (ii) the formation of an imidazolium salt of the type 2[iPr2Im-Ar]+[NiBr4]2- from trans-[Ni(iPr2Im)2(Br)2] 2 and ArBr. The reactions of complex 2 with a series of aryl halides at higher temperatures leads to the decomposition of the bis(carbene) nickel moiety with formation of the the imidazolium salts 2[iPr2Im-Ar]+[NiBr2X2]- (for X = I, Ar = Ph 10 and X = Br, Ar = Ph 11, 4-MeC6H4 12, 4-FC6H4 13, 4-OSi(CH3)3-C6H4 14) in high yields.
S. Dürr, D. Ertler, U. Radius, Inorg. Chem. 2012, 51, 3904–3909.
Abstract
Degradation of white phosphorus (P4) in the coordination sphere of transition metals is commonly divided into two major pathways depending on the Px ligands obtained. Consecutive metal-assisted P-P bond cleavage of four bonds of the P4 tetrahedron leads to complexes featuring two P2 ligands (symmetric cleavage) or one P3 and one P1 ligand (asymmetric cleavage). A systematic investigation of the degradation of white phosphorus P4 to coordinated µ,η2:2 bridging diphosphorus ligands in the coordination sphere of cobalt is presented herein as well as the isolation of each of the decisive intermediates on the reaction pathway. The olefin complex [Cp*Co(iPr2Im)(η2-C2H4)] 1 (Cp* = η5-C5Me5, iPr2Im = 1,3-di-iso-propylimidazolin-2-ylidene) reacts with P4 to give [Cp*Co(iPr2Im)(η2-P4)] 2, the insertion product of [Cp*Co(iPr2Im)] into one of the P-P bonds. Addition of a further equivalent of the CoI complex [Cp*Co(iPr2Im)(η2-C2H4)] 1 induces cleavage of a second P-P bond to yield the dinuclear complex [{Cp*Co(iPr2Im)}2(μ,η2:2-P4)] 3, in which a kinked cyclo-P44- ligand bridges two cobalt atoms. Consecutive dissociation of the N-heterocyclic carbene with concomitant rearrangement of the cyclo-P4 ligand and P-P dissociation leads to the complexes [Cp*Co(µ,η4:2-P4)Co(iPr2Im)Cp*] 4, featuring a P4 chain, and [{Cp*Co(μ,η2:2-P2)}2] 5, in which two isolated P22- ligands bridge two [Cp*Co] fragments. Each of these reactions is quantitative if performed on an NMR scale, and each compound can be isolated in high yields and large quantities.
S. Dürr, B. Zarzycki, D. Ertler, I. Ivanović-Burmazović, U. Radius, Organometallics 2012, 31, 1730–1742.
Abstract
The complexes [(η5-C5R5)Co(iPr2Im)(η 2-C2H4)] (R = H 1; Me 2) were synthesized in good yields via reaction of one equivalent of the N-heterocyclic carbene (NHC) iPr2Im (R2Im = 1,3-di-alkyl-imidazolin-2-ylidene) and the bis(ethylene) complexes [(η5-C5R5)Co(η2-C2H4)2]. These complexes serve as convenient starting materials for chemistry using the [(η5-C5R5)Co(iPr2Im)] complex fragment. The reaction with carbon monoxide leads to the carbonyl complexes [(η5-C5R5)Co(iPr2Im)(CO)] (R = H 3; Me 4) in good to excellent yields. The carbonyl complexes 3 and 4 are very air sensitive and react readily with oxygen in the solid state and in solution. Whereas the cyclopentadienyl substituted complex [(η5-C5H5)Co(iPr2Im)(CO)] 3 decomposes upon reaction with O2 to untractable products, [(η5-C5Me5)Co(iPr2Im)(CO)] 4 yields the structurally characterized cobalt(III) carbonato complex [(η5-C5Me5)Co(iPr2Im)(η2-CO3)] 5. This reaction represents the first example for O2 oxidation of metal bound carbonyl for a 3d transition metal complex. The oxidation is too fast to be monitored by NMR spectroscopy and application of low-temperature time-resolved UV/Vis spectroscopy combined with stopped-flow techniques led to the detection of a possible intermediate. Based on these experiments and computational investigations using density functional theory (DFT) the peroxo acyl complex [(η5-C5Me5)Co(iPr2Im)(k2-C,O-C{O}OO)] B is assumed to be the key intermediate detected. The DFT calculations further reveal that this reaction is strongly exothermic with two kinetic barriers, one for the exothermic addition of O2 to the carbonyl complexes 4 to give the peroxo acyl complex [(η5-C5Me5)Co(iPr2Im)(k2-C,O-C{O}OO)] B, the other for the rearrangement of B to give the carbonato complex [(η5-C5Me5)Co(iPr2Im)(η2-CO3)] 5. The key step for the rearrangement is the formation of CO2 in the coordination sphere of cobalt and the attack of metal bound oxygen at the carbon atom of CO2.
P. Fischer, K. Götz, A. Eichhorn, U. Radius, Organometallics 2012, 31, 1374–1383 (Special Issue: Fluorine in Organometallic Chemistry).
Abstract
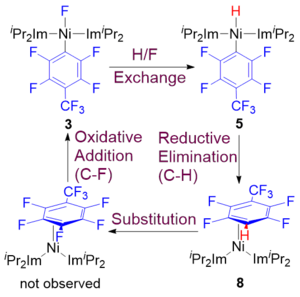
The hydrodefluorination reaction of perfluorinated arenes using [Ni2(iPr2Im)4(COD)] 1 (iPr2Im = 1,3-bis(isopropyl)imidazolin-2-ylidene) as a catalyst as well as stoichiometric transformations to elucidate the decisive steps for this reaction are reported. The reaction of hexafluorobenzene with five equivalents of triphenylsilane in the presence of 5 mol% 1 affords 1,2,4,5-tetrafluorobenzene after 48 hours at 60 °C and 1,4-difluorobenzene after 96 hours at 80 °C; the reaction of perfluorotoluene and five equivalents Et3SiH for 4 days at 80 °C results in the selective formation of 1-(CF3)-2,3,5,6-C6F4H. Stoichiometric transformations of the complexes cis-[Ni(iPr2Im)2(H)(SiPh3)] and cis-[Ni(iPr2Im)2(H)(SiMePh2)] with hexafluorobenzene at room temperature lead to the formation of trans-[Ni(iPr2Im)2(F)(C6F5)] 2 and trans-[Ni(iPr2Im)2(H)(C6F5)] 4 with elimination of the corresponding silane or fluorosilane. The reactions of the C–F activation products trans-[Ni(iPr2Im)2(F)(C6F5)] 2 and trans-[Ni(iPr2Im)2(F)(4-(CF3)C6F4)] 3 with PhSiH3 and Ph2SiH2 afford the hydride complexes trans-[Ni(iPr2Im)2(H)(C6F5)] 4 and trans-[Ni(iPr2Im)2(H)(4-(CF3)C6F4)] 5, which convert into the compounds trans-[Ni(iPr2Im)2(F)(2,3,5,6-C6F4H)] 7, trans-[Ni(iPr2Im)2(F)(3-(CF3)-2,4,5-C6F3H)] 9a and trans-[Ni(iPr2Im)2(F)(2-(CF3)-3,4,6-C6F3H)] 9b, respectively. In the case of the rearrangement of trans-[Ni(iPr2Im)2(H)(4-(CF3)C6F4)] 5 an intermediate [Ni(iPr2Im)2(η2-C,C-(CF3)C6F4H)] 8 was detected. Reaction of 8 with perfluorotoluene gave the C–F activation product trans-[Ni(iPr2Im)2(F)(4-(CF3)C6F4)] 3. All these experimental findings point to a mechanism for the HDF by [Ni(iPr2Im)2] via the “fluoride route” involving C–F activation of the polyfluoroarene, H/F exchange of the resulting nickel fluoride, reductive elimination of the polyfluoroaryl nickel hydride to an intermediate with η2-C,C-coordinated arene ligand, subsequent ligand exchange with the higher fluorinated polyfluoroarene and renewed C–F activation of the polyfluoroarene.
U. Radius, F. M. Bickelhaupt, Coord. Chem. Rev. 2009, 253, 678–686.
Abstract
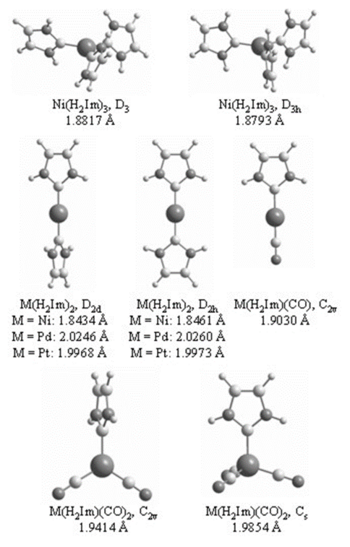
In this contribution, we give a brief overview of studies on the bonding mechanism between transition metals (TM) and N-heterocyclic carbenes (NHC) and report on a systematic bond analysis of the bonding of 1,3-diorganyl-imidazolin-2-ylidenes (R2Im) in a series of nickel, palladium and platinum complexes D2h- and D2d-M(H2Im)2 to exemplify the dependence of the TM-NHC bonding on the group 10 transition metal. Furthermore complexes with seemingly different complex fragment group electronegativities, i.e. [Ni(R2Im)3], [Ni(R2Im)2], [Ni(R2Im)(CO)], [Ni(R2Im)(CO)2], and [Ni(R2Im)(CO)3] have been analyzed, a series that provides theoretical evidence that the bonding mechanism of 1,3-diorganyl-imidazolin-2-ylidene ligands to metal-complex fragments strongly depends on the nature of the ligand environment. Our results confirm the currently accepted idea that NHCs are not pure η-donors. In the series of complexes examined here η-contribution is at least 10% and up to 40%, depending on the transition metal complex fragment bonded to the carbene. The dependence of the bonding mechanism on the R substituent in R2Im also investigated.
T. Schaub, P. Fischer, A. Steffen, T. Braun, U. Radius, J. Am. Chem. Soc. 2008, 130, 9304–9317.
Abstract
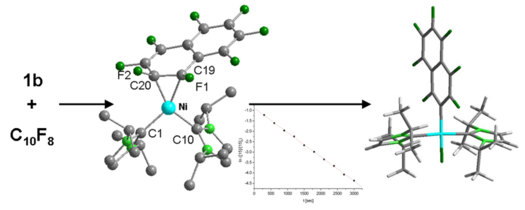
The reaction of [Ni2(iPr2Im)4(COD)] 1a or [Ni(iPr2Im)2(η2-C2H4)] 1b with different fluorinated arenes is reported. These reactions occur with a high chemo- and regioselectivity. In the case of polyfluorinated aromatics of the type C6F5X such as hexafluorobenzene (X = F) octafluorotoluene (X = CF3), trimethyl(pentafluorophenyl)silane (X = SiMe3), or decafluorobiphenyl (X = C6F5) the C-F activation regioselectively takes place at the C-F bond in the para position to the X group to afford the complexes trans-[Ni(iPr2Im)2(F)(C6F5)] 2, trans-[Ni(iPr2Im)2(F)(4-(CF3)C6F4)] 3, trans-[Ni(iPr2Im)2(F)(4-(C6F5)C6F4)] 4, and trans-[Ni(iPr2Im)2(F)(4-(SiMe3)C6F4)] 5. Complex 5 was structurally characterized by X-ray diffraction. The reaction of 1a with partially fluorinated aromatic substrates C6HxFy leads to the products of a C-F activation trans-[Ni(iPr2Im)2(F)(2-C6FH4)] 7, trans-[Ni(iPr2Im)2(F)(3,5-C6F2H3)] 8, trans-[Ni(iPr2Im)2(F)(2,3-C6F2H3)] 9a and trans-[Ni(iPr2Im)2(F)(2,6-C6F2H3)] 9b, trans-[Ni(iPr2Im)2(F)(2,5-C6F2H3)] 10, and trans-[Ni(iPr2Im)2(F)(2,3,5,6- C6F4H)] 11. The reaction of 1a with octafluoronaphthalene yields exclusively trans-[Ni(iPr2Im)2(F)(1,3,4,5,6,7,8-C10F7)] 6a, the product of an insertion into the C-F bond in the 2-position, whereas for the reaction of 1b with octafluoronaphthalene the two isomers trans-[Ni(iPr2Im)2(F)(1,3,4,5,6,7,8-C10F7)] 6a and trans-[Ni(iPr2Im)2(F)(2,3,4,5,6,7,8-C10F7)] 6b in a ratio of 11:1 are formed. The reaction of 1a or of 1b with pentafluoropyridine at low temperatures affords trans-[Ni(iPr2Im)2(F)(4-C5NF4)] 12a as the solely product, whereas the reaction of 1b performed at room temperature leads to the generation of trans-[Ni(iPr2Im)2(F)(4-C5NF4)] 12a and trans-[Ni(iPr2Im)2(F)(2-C5NF4)] 12b in a ratio of approximately 1:2. The detection of intermediates as well as kinetic studies give some insight into the mechanistic details for the activation of an aromatic carbon-fluorine bond at the {Ni(iPr2Im)2} complex fragment. The intermediates of the reaction of 1b with hexafluorobenzene and octafluoronaphthalene, [Ni(iPr2Im)2(η2-C6F6)] 13 and [Ni(iPr2Im)2(η2-C10F8)] 14, have been detected in solution. They convert into the C-F bond activation products. Complex 14 was structurally characterized by X-ray diffraction. The rates for the loss of 14 at different temperatures for the C-F activation of the coordinated naphthalene are first order and the estimated activation enthalpy ΔH¹ for this process was determined to be ΔH¹ = 116 ± 8 kJ mol-1 (ΔS¹ = 37 ± 25 J K-1 mol-1). Furthermore, DFT calculations on the reaction of 1a with hexafluorobenzene, octafluoronaphthalene, octafluorotoluene, 1,2,4-trifluorobenzene, and 1,2,3-trifluorobenzene are presented.
T. Schaub, M. Backes, U. Radius, J. Am. Chem. Soc. 2006, 128, 15964–15965. (Highlighted: Chem. Eng. News 2006, 84(59), 31; Nachr. Chem. 2007, 55, 119)
Abstract
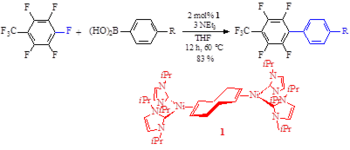
We report here the first example of a catalytically active system for Suzuki-Miyaura type cross-coupling reactions of perfluorinated arenes such as octafluoro toluene and perfluoro biphenyl with aryl boronic acids. This catalysis combines C-F bond activation of these substrates efficiently with C-C coupling reactions. Although C-F activation appears to be quite general in this system, it was necessary to optimize each reaction separately.
T. Schaub, U. Radius, Chem. Eur. J. 2005, 11, 5024–5030.
Abstract
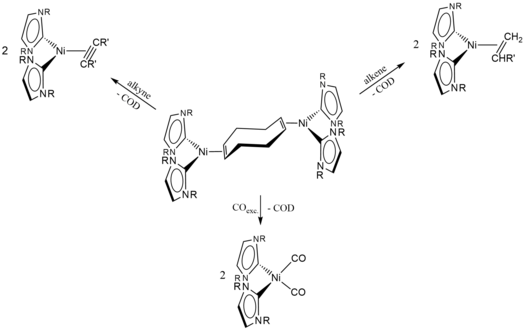
The NHC stabilized complex [Ni2(iPr2Im)4(COD)] 1 was isolated in good yield from the reaction of [Ni(COD)2] with 1,3-di(isopropyl)imidazole-2-ylidene (iPr2Im). Compound 1 is a source of the [Ni(iPr2Im)2] complex fragment in stoichiometric and catalytic transformations. The reactions of 1 with ethylene and CO under atmospheric pressure or with equimolar amounts of diphenyl acetylene lead to the compounds [Ni(iPr2Im)2(η2-C2H4)] 2, [Ni(iPr2Im)2(η2-C2Ph2)] 3, and [Ni(iPr2Im)2(CO)2] 4 in good yields. In all cases the [Ni(iPr2Im)2] complex was readily transferred without decomposition or fragmentation. In the infrared spectrum of carbonyl complex 4, the CO stretching frequencies were observed at 1847 cm-1 and 1921 cm-1, significantly shifted to lower wavenumbers compared to other nickel(0) carbonyl complexes of the type [NiL2(CO)2]. Complex 1 activates the C-F bond of hexafluorobenzene very efficiently to give [Ni(iPr2Im)2(F)(C6F5)] 5. Furthermore, [Ni2(iPr2Im)4(COD)] 1 is also an excellent catalyst for the catalytic insertion of diphenyl acetylene into the 2,2’ bond of biphenylene. The reaction of 1 with equimolar amounts of biphenylene at low temperature leads to the insertion product into the strained 2,2’ bond, [Ni(iPr2Im)2(2,2’-biphenyl)] 6. The reaction of diphenyl acetylene and biphenylene at 80 °C using 2 mol% 1 as a catalyst yields diphenylphenanthrene quantitatively and is completed within one hour.