Pump–probe spectroscopy
Life and all the processes it contains consist of a sequence of events. In order to understand these processes, such as chemical reactions, at the fundamental level of quantum mechanics and possibly to use these processes technologically, we must gain knowledge about the dynamics of molecules and solids, which are composed of atoms. The dynamics of electrons and vibrations in molecules or solids occur on a time scale of femto- to picoseconds (10-15 s to 10-12 s) and is thus many orders of magnitude faster than the switching rates of a few nanoseconds (10-9 s) in modern computer chips. Thus, one needs events to trigger and track the process under investigation which are faster than this process itself.
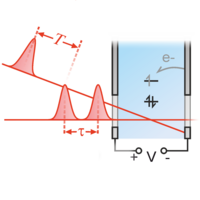
To study ultrafast dynamics in quantum-mechanical systems, we therefore use femtosecond laser pulses, mainly in the optical spectral range. Within these laser pulses, light is concentrated to a short time span of a few femtoseconds, making them suitable as start and stop events in transient absorption spectroscopy, also called "pump–probe spectroscopy".
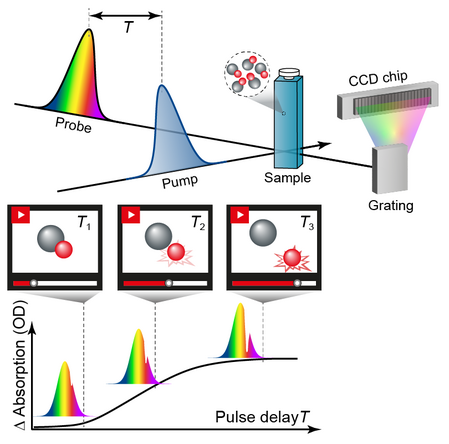
The measuring principle is that a laser pulse, the so-called pump pulse, excites the system under investigation and thus inititiates the dynamics in the system. A second pulse, the so-called probe pulse, interacts with the excited system with a time delay and interrogates its current state. In the above example, excitation with the pump pulse detaches a subsystem, which can then be excited as an independent system in the red spectral region. In transient absorption spectroscopy, an increase in absorption in the red spectral region is then measured as a function of the time delay T between the pump pulse and the probe pulse, which can be determined by the absence of the corresponding spectral components in the probe pulse using a spectrometer. In order to be able to determine the increase or decrease in absorption, the spectra of the probe pulse recorded with and without the presence of the pump pulse are compared.
In our group we develop new methods of transient absorption spectroscopy, which allow to apply the principle of time-resolved spectroscopy to a wide variety of systems and processes. These processes include, for example, the temporal dynamics of chemical reactions.
Chemical processes form the basis of our daily life. Many of these processes and the reactions that take place within them are started by light irradiation. Well-known examples include the photochemical dynamics of receptor molecules in the retina of the eye, which are important for vision, photosynthesis in plants and bacteria, or the formation of vitamin D in human skin under solar radiation. It is therefore obvious that light can also be used as a tool to initiate and study chemical processes in a well-controlled laboratory environment. As an example, the photochemical conversion from the merocyanine isomer TTT to TTC is shown below [1]. Such a conversion finds use in molecular electronics.
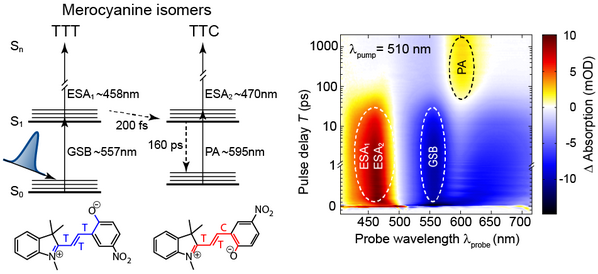
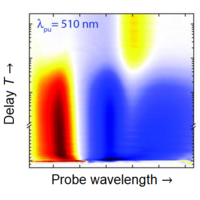
The pump pulse initially transfers many TTT molecules into the electronically excited state S1. Since there are now less TTT molecules in the ground state S0, the absorption of the probe pulse decreases at the corresponding wavelength and the signal becomes negative, i.e., the change in absorption is less than zero (GSB: Ground State Bleach). From the S1 state of TTT, a conversion to the TTC isomer now occurs within 200 fs via coherent dynamics of molecular vibrations. When the excited S1 state of TTC decays to the ground state S0 after 160 ps, the corresponding TTC molecules can be excited again from the S0 to S1 state. Accordingly, the probe pulse at 595 nm will experience more absorption from this species and Δabsorption >0 is the case here. Because this absorption could only occur by the pump-pulse induced isomerization, the signal is also called product absorption (PA). Both isomers, TTT and TTC, also exhibit an excitation by the probe pulse from the respective state S1 to higher excited electronic states Sn shortly after the excitation by the pump pulse. This process is called "excited state absorption" (ESA) and it has a positive signal since the absorption of the probe pulse in the associated spectral region of <500 nm requires prior excitation by the pump pulse [1].
We have developed several new variants of pump–probe spectroscopy, with which we can specifically study, for example, the dynamics of chiral molecules [2] and the dynamics in electrochemical processes [3]. A particular achievement is the possibility to isolate transient absorption spectra of single as well as multiple excitations systematically and background-free, which was not possible for decades [4]. The targeted interrogation of the interaction between multiple excitations thereby enables us to draw conclusions about the energy transport in materials, which are important for technological developments such as solar cells.
References
| [1] | S. Ruetzel, M. Diekmann, P. Nuernberger, C. Walter, B. Engels, and T. Brixner, Here, we use pump–probe spectroscopy with variable excitation wavelength to follow the photoisomerization reaction of a merocyanine dye. |
| [2] | A. Steinbacher, H. Hildenbrand, S. Schott, J. Buback, M. Schmid, P. Nuernberger, and Tobias Brixner, |
| [3] | J. Heitmüller, K. Eckstein, R. Renner, M. Stolte, T. Hertel, F. Würthner, and T. Brixner, In organic solar cells, charge separation is an important step. We have combined an electrochemical cell with femtosecond spectroscopy to study the ultrafast dynamics of charged molecular species. |
| [4] | P. Malý, J. Lüttig, P. A. Rose, A. Turkin, C. Lambert, J. J. Krich, and T. Brixner, We have developed a method to filter out the weak signal of interactions between multiple excitons from the dominant measured signal of absorption from single excitons and demonstrate this method on diverse systems such as polymers, light-harvesting complexes, and nanocrystals. |